
Bài viết này nêu bật một số điểm tương đồng và khác biệt giữa lộ trình quản lý Thiết bị y tế 510(k) và Dấu CE, đồng thời giúp hài hòa một số khía cạnh của chiến lược quản lý tổng thể.
Lưu ý của biên tập viên: Bài viết này cập nhật và thay thế blog năm 2014 của Vincent Crabtree về 510(k) và CE Marking (Pt 1 và Pt 2).
FDA có định nghĩa rõ ràng về lộ trình 510(k), cho phép các nhà sản xuất đưa sản phẩm của họ ra thị trường với tốc độ nhanh hơn và chi phí thấp hơn so với lộ trình Phê duyệt trước khi tiếp thị (PMA) của FDA. Quy trình đánh dấu CE của Liên minh Châu Âu, ban đầu được coi là đơn giản nếu thiết bị được FDA chấp thuận, không còn là một quy trình tầm thường nữa. Việc giới thiệu MDR (2017/745) và IVDR (2017/746) của EU đã khiến một số nhà sản xuất bắt đầu các chương trình khắc phục để cập nhật tài liệu của họ về việc tuân thủ và tiến hành thử nghiệm bổ sung hoặc nghiên cứu lâm sàng.
Báo cáo tóm tắt
Chỉ thị về Thiết bị Y tế (MDD) đang chuyển sang Quy định về Thiết bị Y tế (MDR) ở EU, phân loại lại các thiết bị y tế thành Loại I, Is, Im, Ir, IIa, IIb, III và IIIc (tùy chỉnh). Việc phân loại thiết bị hướng dẫn các yêu cầu về thiết kế, thử nghiệm, xác minh, xác nhận, kiểm tra lâm sàng và giám sát sau khi đưa ra thị trường để thiết bị được đánh dấu CE và đưa vào Thị trường EU.
Không thể so sánh quy trình 510(k) và quy trình Đánh dấu CE trên cơ sở một đối một vì quy trình 510(k) áp dụng cho một số lượng sản phẩm tương đối nhỏ khi so sánh với khả năng áp dụng của quy trình Đánh dấu CE. Những thay đổi đáng kể đối với thiết bị yêu cầu một số mức độ giám sát và báo cáo trong cả hai quy trình và có thể dẫn đến nhu cầu về 510(k) mới hoặc Cơ quan Thông báo phải kiểm tra lại.
Quy trình 510(k) đã phát triển trở nên đơn giản với chương trình eSTAR và chức năng theo dõi tiến trình. Các Chương trình eSTAR hiện đang được thí điểm cho cổng thông tin chung của FDA và Bộ Y tế Canada, điều này sẽ làm cho quá trình này thuận tiện hơn cho các nhà sản xuất. EUDAMED hiện đang được xây dựng và hệ thống hoạt động đầy đủ dự kiến sẽ ra mắt vào quý 3 năm 2024.
Cả hai quy trình 510(k) và CE Marking đều hoạt động theo các bộ quy định khác nhau đối với Hệ thống quản lý chất lượng, nhưng ISO 13485:2016 có thể được sử dụng cùng với việc triển khai bổ sung các điều khoản MDR của FDA và EU. Với đề xuất hài hòa QSR với ISO 13485:2016, việc triển khai Hệ thống quản lý chất lượng sẽ trở nên dễ dàng hơn đối với các nhà sản xuất. Cần phải nộp tài liệu về thiết kế cốt lõi và quản lý rủi ro và tài liệu bổ sung sẽ khác nhau tùy theo thị trường.
Thuật ngữ, sự tương đương và ứng dụng
510(k) là một bản đệ trình trước khi tiếp thị được gửi tới FDA để chứng minh rằng thiết bị được tiếp thị là an toàn và hiệu quả, tức là về cơ bản tương đương (SE), như một thiết bị được tiếp thị hợp pháp. FDA định nghĩa (các) thiết bị được bán trên thị trường hợp pháp mà sự tương đương được coi là “vị ngữ”. Một thiết bị về cơ bản tương đương với một vị ngữ nếu thiết bị chủ thể:
- có cùng mục đích sử dụng như vị ngữ và có cùng đặc điểm công nghệ hoặc các đặc điểm công nghệ khác nhau không đặt ra các câu hỏi khác nhau về an toàn và hiệu quả.
- thông tin được gửi tới FDA chứng minh rằng thiết bị này an toàn và hiệu quả (hoặc hơn) như thiết bị được bán trên thị trường hợp pháp.
Xem Hình 1: Sự tương đương đáng kể theo FDA để biết các bản đệ trình 510(k).
Việc gửi 510(k) được áp dụng cho các thiết bị Loại I, II (trừ khi được miễn) và thiết bị Loại III, nếu có.
Mặc dù mọi quốc gia trong Liên minh Châu Âu, thường được gọi là các Quốc gia Thành viên, đều có Cơ quan có thẩm quyền chịu trách nhiệm tuân thủ các thiết bị liên quan đến chỉ thị đánh dấu CE, nhưng trách nhiệm đánh dấu CE được giao cho Cơ quan Thông báo, để ngăn ngừa xung đột lợi ích, và hài hòa các yêu cầu.
Để có được Dấu CE cho thiết bị, nhà sản xuất phải chứng minh rằng thiết bị của họ tuân thủ các yêu cầu MDR của EU. Thông thường, Cơ quan thông báo có nhiệm vụ xem xét Bản ghi chính của thiết bị và các tài liệu liên quan để phê duyệt Dấu CE cho thiết bị. Một trong những cách thể hiện sự tuân thủ là thông qua sự tương đương. Dấu CE, bằng cách tuyên bố tính tương đương theo MDR của EU, yêu cầu cân nhắc bổ sung để chứng minh tính tương đương. Nhà sản xuất phải công bố sự tương đương về các đặc điểm sau:
- Kỹ thuật – điều kiện sử dụng, thông số kỹ thuật và đặc tính, phương pháp triển khai (nếu có), nguyên tắc hoạt động và yêu cầu hiệu suất quan trọng
- Sinh học - vật liệu hoặc chất tiếp xúc với cùng mô hoặc chất dịch cơ thể của con người, loại và thời gian tiếp xúc tương tự và đặc tính giải phóng của các chất (bao gồm các sản phẩm phân hủy và chất ngâm chiết)
- Lâm sàng – tình trạng hoặc mục đích lâm sàng, mức độ nghiêm trọng và giai đoạn của bệnh, vị trí trên cơ thể, dân số, người sử dụng, hoạt động quan trọng xét đến tác dụng lâm sàng dự kiến cho một mục đích cụ thể đã định.
Việc chứng minh sự tương đương theo Dấu CE đòi hỏi nhiều nỗ lực hơn và đôi khi thông tin độc quyền có thể không có sẵn để cố gắng khẳng định sự tương đương với các sản phẩm của một nhà sản xuất khác. Không cần phải có sự phê duyệt của Cơ quan Thông báo đối với Dấu CE đối với Loại I theo MDR của EU. Các thiết bị loại I được tự chứng nhận theo MDR của EU. Các thiết bị loại I được cung cấp dưới dạng vô trùng (Is), có chức năng đo lường (Im) hoặc là dụng cụ phẫu thuật có thể tái sử dụng (Ir) phải tuân theo Dấu CE.
 và Dấu CE")
Hình 1: Sự tương đương đáng kể theo FDA đối với các bản đệ trình 510(k).
Thay đổi đối với thiết bị
Các thiết bị trải qua thay đổi thiết kế có thể yêu cầu gửi 510(k) mới. Phải nộp 510(k) mới trong các trường hợp sau:
- Những thay đổi được thực hiện nhằm mục đích ảnh hưởng đáng kể đến sự an toàn hoặc hiệu quả của thiết bị.
- Những thay đổi chính về ghi nhãn – Thêm chống chỉ định, dán nhãn lại các thiết bị là có thể tái sử dụng sau một lần sử dụng, v.v.
- Những thay đổi lớn về công nghệ, kỹ thuật và hiệu suất – những thay đổi trong cơ chế điều khiển, nguyên lý vận hành, thay đổi loại năng lượng, v.v.
- Thay đổi vật liệu
- Các sửa đổi dẫn đến thay đổi đáng kể trong hồ sơ rủi ro của thiết bị.
Mặc dù danh sách trên có vẻ đơn giản nhưng có một số trường hợp trong đó ranh giới bị mờ và thiết bị có thể không yêu cầu 510(k) mới. Hướng dẫn của FDA Quyết định thời điểm gửi 510(k) để thay đổi thiết bị hiện có cung cấp sơ đồ chi tiết hỗ trợ việc ra quyết định gửi 510(k) mới. Một hướng dẫn riêng thảo luận về thời điểm thay đổi phần mềm yêu cầu một 510(k) mới.
Theo MDR EU, các thiết bị trải qua bất kỳ thay đổi đáng kể nào về một hoặc nhiều danh mục sau đây phải báo cáo thay đổi đó cho Cơ quan thông báo đã chứng nhận thiết bị.
- chủ đích
- đặc điểm kỹ thuật thiết kế hoặc hiệu suất
- thành phần hoặc vật liệu
- khử trùng hoặc thiết kế bao bì có tác động đến việc khử trùng
- phần mềm
Định nghĩa về sự thay đổi đáng kể có thể mơ hồ. Hướng dẫn MDCG cung cấp thông tin bổ sung về những thay đổi quan trọng theo MDR của EU. Cơ quan thông báo có thể quyết định đánh giá lại QMS hoặc tài liệu kỹ thuật của nhà sản xuất, nếu cần.
Một chiến lược quản lý tốt sẽ bao gồm cả các yêu cầu MDR của FDA và EU để đảm bảo rằng cả hai đều được đáp ứng.
Quy trình và Phí
Việc gửi 510(k) có thể được thực hiện trực tuyến bằng cổng CDRH. Cổng CDRH cho phép các nhà sản xuất gửi bản gửi Thiết bị y tế bằng eSTAR, một biểu mẫu PDF tương tác. Cổng CDRH cũng cung cấp trình theo dõi tiến trình hiển thị trạng thái gửi. Cổng có một số hạn chế về kích thước và loại tệp nhưng cho phép phóng viên gửi tài liệu quá khổ qua thư đến Trung tâm Kiểm soát Tài liệu CDRH (DCC). Bất kỳ thiết bị nào đã được cấp giấy phép 510(k) đều có thể được đưa ra thị trường ngay lập tức trong khi chờ kiểm tra Hệ thống Chất lượng FDA (21 CFR 820), việc kiểm tra này có thể diễn ra sau khi giấy phép.
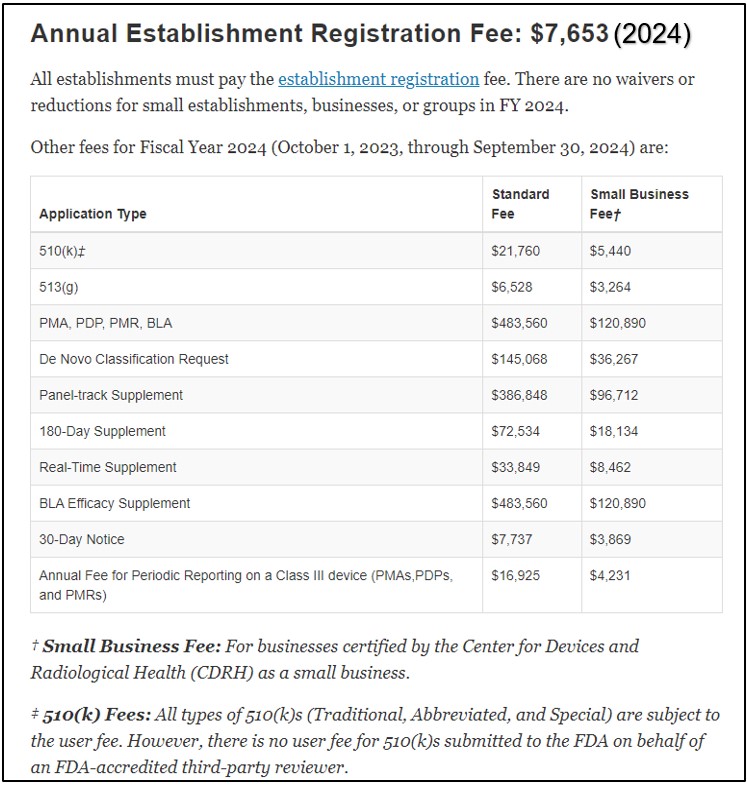
Chi phí đăng ký cho 510(k) được công bố trên trang web của FDA dưới Sửa đổi phí người dùng thiết bị y tế (MDUFA). Lệ phí khác nhau đối với các doanh nghiệp tiêu chuẩn và doanh nghiệp nhỏ. Bảng dưới đây cung cấp ý tưởng về cấu trúc phí. Lệ phí được cập nhật hàng năm tài chính. Mọi cơ sở đều phải đóng phí hàng năm lệ phí đăng ký thành lập.
Quy trình Đánh dấu CE yêu cầu sự tham gia của Cơ quan thông báo và Kiểm tra QMS đủ điều kiện trước khi thiết bị có thể được đánh giá để lấy Dấu CE. Chọn Cơ quan thông báo của bạn dựa trên sự kết hợp phù hợp giữa phí, sắp xếp chuyến đi và kiến thức chuyên môn phù hợp với bạn.
MDR EU đã đưa ra khái niệm về cơ sở dữ liệu Cơ sở dữ liệu Châu Âu về Thiết bị Y tế (EUDAMED) nhằm cung cấp “bức tranh sống động về vòng đời của các thiết bị y tế được cung cấp tại Liên minh Châu Âu (EU)”. EUDAMED sẽ bao gồm sáu mô-đun liên quan đến: đăng ký tác nhân, nhận dạng thiết bị duy nhất (UDI) và đăng ký thiết bị, Cơ quan thông báo và chứng chỉ, điều tra lâm sàng và nghiên cứu hiệu suất, cảnh giác và giám sát thị trường. Tính đến tháng 2024 năm 3, EUDAMED có ba mô-đun trực tiếp – người vận hành kinh tế, thiết bị và chứng chỉ. Ba mô-đun cuối cùng dự kiến sẽ được đưa lên mạng vào quý 2024 năm XNUMX.
Trong trường hợp không có hệ thống trực tuyến để quản lý việc gửi CE Marking, nhà sản xuất phải sử dụng cổng/thư mục chia sẻ/các phương pháp khác do Cơ quan thông báo thiết lập để chia sẻ tài liệu và bằng chứng cần thiết để chứng nhận. Cơ quan thông báo tiến hành Kiểm tra QMS theo các yêu cầu của Hệ thống quản lý chất lượng MDR của EU (Phụ lục IX) trước khi chứng nhận thiết bị cho thị trường. Các thiết bị không thể được đánh dấu CE và đưa ra thị trường nếu không có đánh giá tài liệu kỹ thuật và kiểm toán QMS của Cơ quan Thông báo (hoặc lấy mẫu trong một số trường hợp), trừ khi thiết bị được phân loại là Loại I.
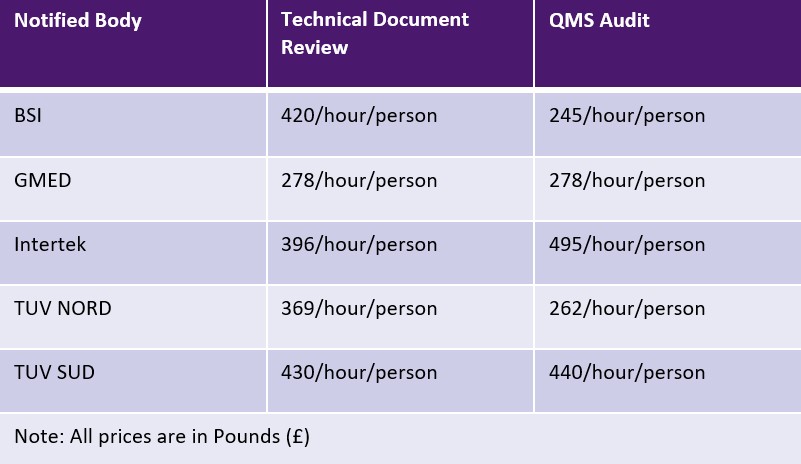
MDR của EU cho phép Cơ quan thông báo tính phí cố định hoặc phí theo thời gian cho mỗi hoạt động. Tuy nhiên, Cơ quan Thông báo không bị ràng buộc bởi bất kỳ giới hạn nào đối với các khoản phí. Bảng dưới đây trình bày mẫu các Cơ quan Thông báo và phí của họ. Có thể áp dụng phí bổ sung cho việc đi lại và chuyên môn.
Hệ thống quản lý chất lượng
Các nhà sản xuất phải thực hiện thẩm định và đảm bảo rằng Hệ thống quản lý chất lượng phù hợp được áp dụng trước khi đưa thiết bị của họ ra thị trường.
FDA quản lý hệ thống quản lý chất lượng theo Quy định hệ thống chất lượng (QSR) (21 CFR 820), áp dụng cho các nhà sản xuất có ý định phân phối thương mại các thiết bị y tế. Một quy tắc mới đã được thông qua để kết hợp Hệ thống quản lý chất lượng – Thiết bị y tế ISO 13485:2016 trong QSR nhằm nỗ lực hài hòa các yêu cầu trên diện rộng. ISO 13485:2016 được nhiều cơ quan quản lý khác trên thế giới sử dụng làm tiêu chuẩn cho Hệ thống quản lý chất lượng. Mặc dù tính linh hoạt của thông quan 510(k) cho phép các nhà sản xuất đưa thiết bị của họ ra thị trường nhưng chúng có thể bị FDA kiểm tra bất cứ lúc nào. Do đó, lợi ích tốt nhất của nhà sản xuất là đảm bảo áp dụng Hệ thống quản lý chất lượng tuân thủ trước khi đưa thiết bị ra thị trường.
Với MDR của EU, ISO 13485:2016 không phải là yêu cầu đối với Dấu CE. Hệ thống quản lý chất lượng được áp dụng phải tuân thủ các quy định nêu tại Phụ lục IX. Các nhà sản xuất phải hoàn thành đơn đăng ký đánh giá Hệ thống quản lý chất lượng của họ với Cơ quan thông báo. Ngoài việc kiểm tra Hệ thống quản lý chất lượng, Cơ quan thông báo sẽ đánh giá tài liệu kỹ thuật của thiết bị dựa trên Hệ thống quản lý chất lượng để đảm bảo rằng tất cả các yêu cầu đều được đáp ứng trước khi được chứng nhận.
Để hài hòa Hệ thống quản lý chất lượng, cách tốt nhất là triển khai hệ thống tuân thủ ISO 13485:2016 và Chương trình kiểm tra đơn thiết bị y tế (MDSAP) cùng với các yêu cầu tuân thủ bổ sung đối với FDA, EU MDR, Bộ Y tế Canada hoặc các quốc gia khác nơi nhà sản xuất dự định kinh doanh .
Tài liệu
FDA và EU MDR yêu cầu một quy trình thiết kế có kiểm soát đối với các thiết bị.
Việc gửi 510(k) yêu cầu Tệp lịch sử thiết kế (DHF) hoàn chỉnh nêu chi tiết các yêu cầu hệ thống, kiến trúc, thông số kỹ thuật, xác minh và xác thực cũng như tài liệu về các hoạt động quản lý rủi ro. Cấu hình 510(k) eSTAR cho phép nhà sản xuất đính kèm tài liệu liên quan vào từng phần của ứng dụng. FDA cung cấp một Danh sách kiểm tra chấp nhận để hướng dẫn nhà sản xuất quy trình 510(k) và đảm bảo có sẵn tài liệu phù hợp.
Đối với MDR EU, Tệp kỹ thuật chứa các tài liệu DHF và cũng yêu cầu nhà sản xuất cung cấp danh sách kiểm tra bổ sung sau:
- Yêu cầu về An toàn và Hiệu suất Chung (GSPR) theo Phụ lục I, trước đây gọi là Danh sách kiểm tra Yêu cầu Thiết yếu theo MDD
- Tài liệu kỹ thuật tiêu chuẩn (SteD) theo Phụ lục II
- Giám sát sau thị trường (PMS) theo Phụ lục III
Tài liệu kỹ thuật cấp cao thường tham chiếu đến các tài liệu DHF khác nhau và danh sách kiểm tra.
Trong cả hai trường hợp, cần phải có tệp quản lý rủi ro đầy đủ tuân thủ ISO 14971.
Tài liệu tham khảo:
- Thông báo tiếp thị trước 510(k) | FDA
- Chương trình 510(k): Đánh giá sự tương đương đáng kể trong các thông báo trước khi tiếp thị [510(k)] (fda.gov)
- Quyết định thời điểm gửi 510(k) để thay đổi thiết bị hiện có - Hướng dẫn dành cho nhân viên ngành và Cục quản lý thực phẩm và dược phẩm (fda.gov)
- Quyết định thời điểm gửi 510(k) cho việc thay đổi phần mềm đối với thiết bị hiện có - Dự thảo hướng dẫn dành cho nhân viên ngành và Cục quản lý thực phẩm và dược phẩm (fda.gov)
- MDCG 2019-15, Ghi chú hướng dẫn dành cho nhà sản xuất thiết bị y tế loại I
- Gửi và theo dõi các bản gửi tiếp thị trước thiết bị y tế trực tuyến: Cổng thông tin CDRH | FDA
- Cơ sở dữ liệu EUDAMED – EUDAMED (europa.eu)
- Sửa đổi phí người dùng thiết bị y tế (MDUFA) | FDA
- MDCG 2023-2, Danh sách phí tiêu chuẩn
- Quy định về Hệ thống Chất lượng (QS)/Thực hành Sản xuất Tốt Thiết bị Y tế | FDA
- Danh sách kiểm tra chấp nhận cho 510(k)s | FDA
- Chương trình eSTAR | FDA
- FDA Health Canada eSTAR (starfishmedical.com)
hình ảnh: Cổ phần của Adobe & StarFish y tế
Dhruvitha Krishna là một Chuyên gia QA/RA tại StarFish Medical với bằng Thạc sĩ Kỹ thuật Y sinh. Cô đã từng làm việc trong lĩnh vực sản xuất, triển khai sản phẩm mới, triển khai phần mềm, quản lý dự án và lĩnh vực pháp lý trong các công ty thiết bị y tế. Dhruvitha luôn nỗ lực cải tiến chất lượng, quy định và liên tục cho nhà sản xuất.
- Phân phối nội dung và PR được hỗ trợ bởi SEO. Được khuếch đại ngay hôm nay.
- PlatoData.Network Vertical Generative Ai. Trao quyền cho chính mình. Truy cập Tại đây.
- PlatoAiStream. Thông minh Web3. Kiến thức khuếch đại. Truy cập Tại đây.
- Trung tâmESG. Than đá, công nghệ sạch, Năng lượng, Môi trường Hệ mặt trời, Quản lý chất thải. Truy cập Tại đây.
- PlatoSức khỏe. Tình báo thử nghiệm lâm sàng và công nghệ sinh học. Truy cập Tại đây.
- nguồn: https://starfishmedical.com/blog/medical-device-510k-ce-marking/

