Integrating reverse transcription in caPCR
We first introduced an RT step to the standard caPCR assay. caPCR is performed using a set of 4 heaters that clamp on either face of a cartridge containing a toroidal chamber filled with the PCR reaction mixture4. By setting heaters on one side of the chamber to the PCR annealing temperature and the other to the denaturation temperature, a gradient is established, thereby producing a passive convective flow that drives PCR amplification. The pre-quenched toehold probe array is immobilized on the surface of the cartridge and responds through TMSD as specific PCR products are synthesized. To accommodate an initial reverse transcription step, we programmed the heaters to maintain a constant temperature of 42 °C for cDNA synthesis for 10 minutes, after which the heaters adjust to 57 °C and 95 °C respectively to initiate convective flow (Fig. 2a). A one-pot RT-PCR approach is used to reduce the number of manual handling steps in the workflow. Toehold probe-mediated detection on the caPCR platform functions optimally under asymmetric PCR conditions: an excess of the primer that produces the strand which displaces the quencher generates a stronger signal on the fluorescent probe array than a 1:1 forward-to-reverse primer ratio (Fig. S1). We ensured that this same primer also mediates cDNA synthesis from RNA targets to avoid the need to introduce separate primers for reverse transcription and PCR stages.
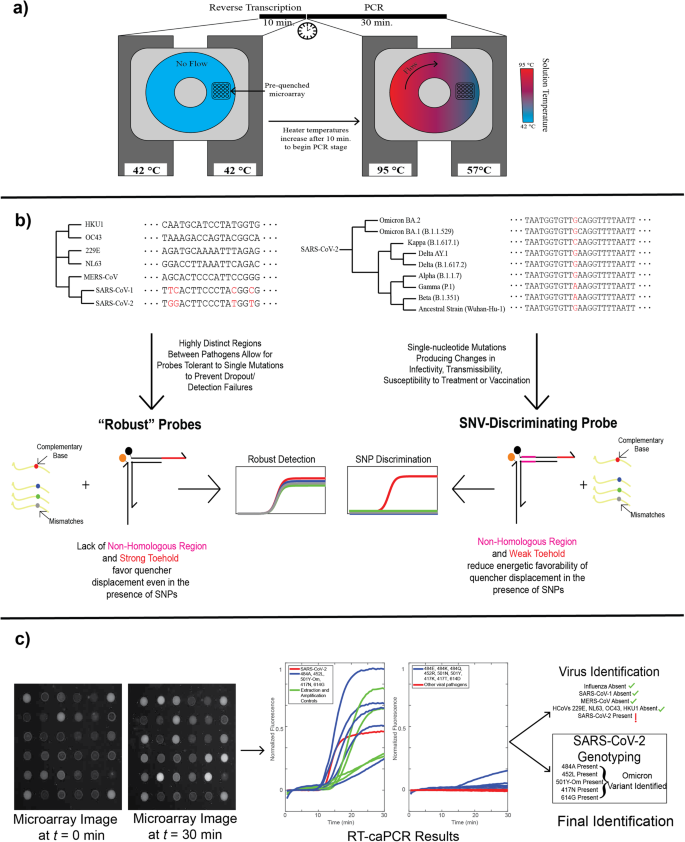
a The caPCR workflow can be extended to include a reverse transcription step (RT-caPCR) through the addition of a reverse transcriptase enzyme, and a dedicated RT stage with all device heaters set to the RT temperature (42 °C). Heater temperatures are then increased to different temperatures for denaturation (95 °C) and annealing (57 °C), inducing convective flow and PCR amplification of the cDNA templates. b The energetic design scheme of probes, based on DNA binding thermodynamics, can be used to address two needs in infectious disease diagnostics: I) Viral pathogen detection should be robust to small changes in the nucleotide sequence to prevent dropout if new variants arise. II) Variant discrimination requires single-nucleotide resolution. Variation of the toehold, domain, and non-homologous region (NHR) energies can be used to meet both of these requirements. c Example data output from a donut RT-caPCR run. Probe array images are acquired every 30 seconds for 30 minutes of caPCR. Fluorescence traces are extracted and analyzed to identify which respiratory pathogens are present and, if applicable, what variant of SARS-CoV-2 is present.
To develop tunable probes with programmable sequence selectivity, ranging from mutation-sensitive to mutation-tolerant, we utilized 2 sets of energetic criteria for toehold probes (Fig. 2b). Robust probes feature a stronger toehold region and a correspondingly weaker domain to reduce the overall influence of a single mutation on the initiation of the binding reaction and subsequent quencher arm displacement. For SNV discrimination, the toehold is reduced in length, and a non-homologous region (NHR) lacking complementarity to the target is introduced. This combination serves to reduce the overall energetic favorability for toehold binding and quencher arm displacement, thereby providing extra weight to single mismatches in the target sequence.
We applied RT-caPCR with both robust and SNV-discriminating probes to respiratory virus detection. Probe array images from the RT-caPCR system are processed using a custom MATLAB script to produce fluorescence traces which are then interpreted to identify both the viral target(s) present and, if applicable, the variant of SARS-CoV-2. An example is shown for the SARS-CoV-2 Omicron variant in Fig. 2c. Thus, our one-pot RT-caPCR assay demonstrates its ability to simultaneously perform both sensitive detection with robust probes as well as single-nucleotide resolution with SNV-discriminatory probes to accurately identify RNA targets of interest in a single reaction.
Fine-tuning the energetics between robust and SNV-discriminating probes
To increase the diversity of targets detected in our RT-caPCR assay, we optimized the energetics of toehold binding and strand displacement to modulate the sensitivity-specificity tradeoff. As a case study, we used the SARS-CoV-2 ‘484E’ genotype as the template for our energetic assessment. Mutations at this site are associated with reduced neutralizing activity from host antibodies12, and 3 of 4 possible nucleotide bases (adenine, guanine, and cytosine) are found in the first position of the codon in naturally evolved SARS-CoV-2 variants9. Uracil substitution at this site would produce a stop codon and thus has not been observed in nature, but for rigor we included a thymine condition in our assessment. We designed a probe for each of the 4 possible nucleotides at this locus and established 6 different thermodynamic conditions based on the binding energy (ΔGo) of the 3 regions of the toehold probe (NHR, domain, and toehold; Fig. 3a, see Materials and Methods). These energetic conditions range from the most specific (Probe 1), in which the energetics of the NHR and toehold are approximately equal (around −9 kcal/mol), to the most sensitive (Probe 6), in which there is no NHR and the toehold is slightly stronger (−12 kcal/mol). In all cases, the domain binding energy is adjusted to keep the overall probe binding energy constant at approximately −40 kcal/mol.
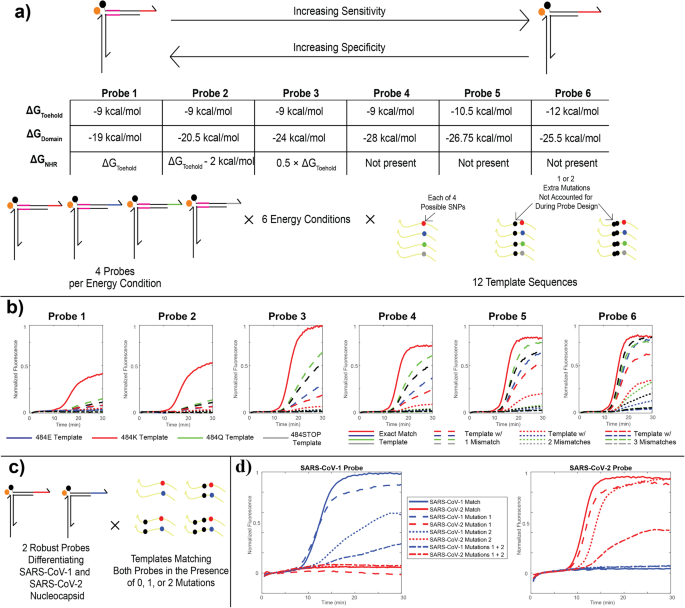
a Design parameters for probe energetic characterization. Six different energetic schemes were designed by varying the binding energies of the toehold, domain, and NHR regions, to permit a tradeoff between single-nucleotide specificity and sensitivity in the form of robustness to mutations. Four different probes were designed for each of the six energetic designs, covering all four possible codons at the 484 mutation site in the SARS-CoV-2 spike protein, three of which have been observed in variants. Each probe was tested with 12 different plasmid templates, including the 4 unique single-nucleotide mutations alone and in the presence of one or two other mutations found further upstream in the Omicron variant. b caPCR results from the 484 K series of probes. Red solid curves denote the on-target performance with no mutations. Dashed, dotted, and dash-dot lines denote templates with 1, 2, or 3 mismatches relative to the probe sequence, respectively. The color specifies the codon at the 484 site (E, K, Q, or STOP). c Scheme for testing robust SARS-CoV-1/2 discrimination. Two probes were designed according to the energetic scheme “Probe 5” in part a for a region of the nucleocapsid gene in SARS-CoV-1 and SARS-CoV-2. Both probes were tested in the presence of the exact-match template and variants including one or both of two upstream mismatches, as well as in the presence of the exact-match template and mismatched variants of the other probe. d Results of the tests described in (c). Solid, dashed, dotted, and dash-dot lines denote exact match templates, templates with either of two SNVs, or templates with both SNVs relative to the SARS-CoV-1 (blue) or SARS-CoV-2 (red) genome.
We tested these probes with 12 different DNA templates, 4 of which represent the exact-match template for each of the 4 possible bases in the 484 codon. To demonstrate the tunability of our probe energetics, we took mutations that arose in later lineages of SARS-CoV-2 evolution and introduced them into the templates: the T478K mutation found in the Delta AY and Omicron lineages, and the S477N mutation found in Omicron lineages (schematic Fig. 3a)10. All 24 probes were evaluated in response to the amplification of each of the 12 templates. Figure 3b shows a subset of these results, specifically for the probes designed to target the 484 K mutation found in Beta and Gamma VOCs10. As the strength of the NHR is reduced and the toehold increases, detection for the exact-match template (solid red line) remains strong and the probe becomes more sensitive to templates with 1 (dashed lines) and 2 (dotted lines) mismatches. This demonstrates our ability to engineer probe energetics for very specific target-binding outcomes. Based on these results, we selected energetic scheme 2 for SNV-discriminating probes and scheme 5 for robust probes.
We designed 2 robust probes targeting the nucleocapsid gene sequence of SARS-CoV-1 and SARS-CoV-2. To ensure that these pathogen sequences could be differentiated using robust probes while remaining tolerant to mutations, we devised 8 templates containing either no mismatches, 1 of 2 mismatches, or both mismatches together (Fig. 3c, see Materials and Methods). The region selected for SARS-CoV-1 and SARS-CoV-2 detection differs between the 2 target sequences by 8 mutations: as such, mismatches in the template were selected based on these differences. As such, the SARS-CoV-2 template with 2 mismatches only has 6 differences relative to the SARS-CoV-1 sequence, thus highlighting a real-life application of the importance for precise tunability of probes for sensitivity versus specificity. Figure 3d shows the results for these tests. In all cases, we observed no cross reactivity between the SARS-CoV-1 and SARS-CoV-2 probes and templates: SARS-CoV-1 RNA was only detected by the SARS-CoV-1 probe, and SARS-CoV-2 RNA was only detected by the SARS-CoV-2 probe. The templates containing either of the 2 mutations can also be unambiguously detected by each appropriate probe, with 1 of the 2 mutations more clearly affecting performance than the other. Similarly, templates with both mutations together are still detected though performance is slightly compromised due to less energetic favorability in quencher arm displacement.
Through our targeted studies of probe energetics, we demonstrated our capacity to precisely engineer probes to have the desired tradeoff of sensitivity versus specificity. SNV-discriminatory probes have energetics that require an exact match for displacement of the quencher arm, while robust probes are more promiscuous in their binding of targets with the ability to detect an amplicon containing up to two mutations. The energetic range we explored can be applied to adjust probes as needed from energy scheme 1 to energy scheme 6 to allow for detection of targets with the desired number of mismatches from the probe sequence.
Performance of robust and SNV-discriminatory probes
To demonstrate proof-of-concept for our highly tunable probe design, we created a full panel of both robust and SNV-discriminatory probes. Our panel highlights the utility of robust probes by screening for all 7 coronaviruses known to infect humans (HCoVs 229E, HKU1, NL63, and OC43; MERS-CoV, SARS-CoV-1, and SARS-CoV-2) as well as influenza A and B8. Additionally, our panel demonstrates the functionality of SNV-discriminatory probes by using 14 probes and 3 amplicons covering 5 commonly mutated sites in the SARS-CoV-2 spike gene to permit differentiation amongst variants of concern (417, 452, 484, 501, and 614; Fig. S2)9,10,11.
To assess the performance of the robust probes in our RT-caPCR assay for sensitive target detection, we performed 65 RT-caPCR tests using coronavirus-negative viral transport media (VTM) samples covering a range of patient demographics (Tables S1–S4) spiked with synthetic RNA from each of the 7 human coronaviruses, either purchased from Twist Bioscience or synthesized in-house using in vitro transcription (IVT) from gene fragments (Table S5). RNA was isolated and purified from these contrived clinical samples using a bead-based extraction method13 due to the precedence for using such methodology for RNA extraction in SARS-CoV-2 diagnostic workflows14 and was used as input to our system. This assay also included both an extraction control (RNA from MS2 bacteriophage spiked into the contrived sample prior to purification) and an amplification control (internal positive control (IPC) RNA spiked into the RT-caPCR reaction mix). Unless otherwise specified, experiments here received approximately 106 copies of MS2 RNA and 107 copies of viral RNA. The RT-caPCR reaction was performed in 40 minutes using 5,000 molecules of IPC control RNA, the extracted sample RNA, and 11 primer pairs covering all targets and controls.
RT-caPCR data were analyzed to identify the viral targets present in each sample, and, in the case of SARS-CoV-2, which specific variant was present (Fig. 4). Figure 4a shows a representative run with the SARS-CoV-2 Omicron variant as the RNA target. All robust probes for viral discrimination are shown in red while SNV-discriminating probes are shown in blue. The extraction and amplification controls are shown in green. Positive and negative probe spots correspond to fully unquenched or fully quenched probes using sequences for which no primers nor template were included and are shown in gray. This type of data can be summarized in a compressed plot as shown in Fig. 4b: all expected positive curves are depicted on the left-hand plot, and all expected negative curves are shown on the right-hand plot. While there is a small level of off-target activity on some SNV-specific probes, comparing the on-target SNV-specific probe curves (484 A, 452 L, 501Y-Om, 417 N, and 614 G) with other SNV-specific probes confirms that the on-target probes respond most strongly in all cases, as assessed by eye as well as through our fluorescence derivative-based SNV-discrimination algorithm (see Materials and Methods and Supplemental Methods). We note that, due to the Omicron variant’s high mutational load relative to other variants11, we designed the sequence of the 484 A probe to also include S477N and T478K mutations found in this variant to improve detection10. Similarly, a unique 501Y-Om probe includes Q493R, G496S, and Q498R mutations that are specifically found in the Omicron variant. A probe specific for the BA.2 subvariant of the Omicron lineage (501Y-Om2) was introduced later. This probe is identical to the 501Y-Om sequence but lacks the G496S mutation10.

a Example results from an RT-caPCR run detecting the SARS-CoV-2 Omicron variant of concern. Robust viral detection probes are in red, SNV-discriminating probes are in blue, extraction and amplification controls are in green, and positive and negative controls are in gray. Each curve represents the intensity of the corresponding probe array spot at each time point over the 60 acquired images, normalized to the brightest spot in the array. b Summary results corresponding to the run in a. All expected positives (extraction and amplification controls, on-target viral identification, and all on-target SNV-discriminating probes) are shown in the left-hand plot, whereas all expected negative probes (all other viral and SNV-discriminating probes, as well as the negative controls) are shown in the right-hand plot. c Viral detection results for human coronavirus 229E, Middle East Respiratory Syndrome coronavirus (MERS-CoV), and SARS-CoV-1 in the format described in (b). d. SNV-discrimination results from a SARS-CoV-2 Delta variant template in the format specified above.
Figure 4c shows compressed representative results from 3 runs for 3 different viral pathogen templates: human coronavirus 229E, MERS-CoV, and SARS-CoV-1. All 3 samples show clear detection of the viral RNA target with the robust pathogen identifying probes, no off-target viral identification probe activity, and the presence of all controls.
Table 1 summarizes the viral identification results from all valid trials. Competition for PCR reaction resources (e.g., polymerase, dNTPs) may suppress amplification of lower copy number targets, so an assay run was considered valid if either a viral pathogen was detected and at least 1 of the 2 controls (MS2 and IPC) was detected, or no viral pathogen was detected but both controls were detected. 11 of the samples were negative controls, which included only MS2 and IPC control RNAs. In all 11 of these samples, both IPC and MS2 were detected (100%). 36 of 36 SARS-CoV-2 samples were identified (100%) as assessed by detection of the SARS-CoV-2 nucleocapsid probe. The remaining human coronaviruses (229E, NL63, OC43, and HKU1) as well as SARS-CoV-1 and MERS-CoV targets also show high sensitivity, with 100% detection for all targets. Both the process of primer binding and amplicon-mediated quencher displacement of the probes contribute to the specificity of the panel, which was 100% across all targets. Here, specificity is calculated as the proportion of true negatives correctly detected. In short, no template induced detection at a robust probe other than the one for which it was specifically designed.
Figure 4d shows a representative run highlighting the ability of our RT-caPCR assay to both identify the RNA target as well as provide single-nucleotide resolution by showing viral variant detection concurrently with pathogen identification for the SARS-CoV-2 Delta variant. While the 452 L probe shows significant off-target activity, its detection is clearly less efficient than the on-target 452 R probe (the blue curve with the highest intensity in the left-hand plot). Thus, SNV-discriminating probes allow for clear distinction between SARS-CoV-2 variants that differ only by a few single mutations.
Table 2 summarizes the performance of the SNV-specific probes for SARS-CoV-2 variants across 36 trials. The number of expected detections is equal to the number of runs for which a template displaying that mutation was included, while the number of expected negatives is the number of runs for which the corresponding template was not included. True positives are runs for which the probe of interest was the dominant SNV-specific probe for that mutation site (as assessed algorithmically), and true negatives are those runs for which it was not. In one run, we note that the 614 G probe was obscured by a foreign particle (dust), so the total for these targets is 35, rather than 36. Aside from 484E, all probes show at least 90% sensitivity. Poor performance of the 484E probe can be partially explained by the presence of a SNV (T478K mutation) in the amplicon for the Delta variants. All other probes show 100% specificity, as an amplicon will always preferentially displace its matching quencher over one with a mutation, assuming the on-target energetics are equal.
Together, these data highlight the ability of robust probes and SNV-discriminatory probes to work in tandem to quickly identify the RNA target present (with the robust probe) and provide single-nucleotide resolution (with the SNV-discriminatory probe).
Evaluating the limit of detection of RT-caPCR
We next performed a series of experiments to determine the limit of detection of our RT-caPCR assay. In all previous tests in Fig. 4, a viral input of 107 molecules was spiked into the VTM prior to extraction, similar to estimates of peak viral load for Omicron and Delta patients15,16. Here we quantified the LOD of our assay by varying the number of input copies of viral RNA and keeping the control copies the same. Using purified RNA either obtained from Twist Bioscience or synthesized in-house from gene fragments via IVT, we achieved a 1,000-molecule LOD for all 7 coronaviruses. Representative results are shown in Fig. S3. In both Fig. S3a and S3b, we find that we can successfully detect down to 1,000 molecules per reaction for a single target with both controls (HCoV 229E; Fig. S3a) and for a SARS-CoV-2 variant with both controls (Omicron; Fig. S3b), a quantity 3 orders of magnitude below that used for VTM testing when using purified RNA. When only the presence of a single target is varied, as in Fig. S3a, we find a clear delay in threshold time for each order of magnitude for only this target, whereas the 2 controls are consistent across runs. This trend is less clear for SARS-CoV-2, which is to be expected given that this target requires simultaneous amplification of 4 amplicons and 2 controls. Competition for enzymatic resources and dNTPs appears to disrupt the clear trend observed for single-amplicon targets. In Fig. S3c, a dilution series was performed by varying the amount of input RNA to the extraction pipeline for the Omicron variant. Here, too, we see detection down to the lowest tested value (10,000-molecule input into the RNA extraction step with roughly 1,000 molecules going into the RT step due to losses during extraction and not using the full 25 µL elution volume in the reaction) and an expected delay in amplification as a function of input concentration.
Detection of multiple targets in a single sample
Having demonstrated the success of our tunable-probe RT-caPCR assay at identifying RNA targets with robust probes (each virus) with single nucleotide specificity with SNV-discriminating probes (SARS-CoV-2 variants), we next explored how our assay performed when assessing more complex inputs by simulating co-infection samples containing multiple RNA targets. Here, an input amount of 105 molecules of each template was spiked directly into the RT-caPCR reaction with no VTM extraction steps. We also expanded our RT-caPCR assay to test for more RNA targets by including 3 additional primer sets and 4 probes to test for influenza A and B (2 targeting the M1 matrix protein gene of influenza A17 and 2 targeting nonstructural protein-1 (nsp1) in the Yamagata and Victoria lineages of influenza B18). This 14-plex assay was tested for all 4 new targets to confirm its efficacy (Fig. S4).
We assessed each of 3 different viral templates alone as well as in 3 different combinations: SARS-CoV-2 Delta variant + HCoV229E (Fig. 5a), SARS-CoV-2 Delta variant + FluB Yamagata lineage (Fig. 5b), and SARS-CoV-2 + HCoV229E + FluB Yamagata lineage (Fig. 5c). Data is presented as compressed fluorescent trace images as described above with expected positive probes in the left panel and expected negative probes in the right panel. Across all runs, we observed the expected behavior: robust (virus identification) probes report the presence of all pathogen templates included, and SNV-discriminating probes (variant identification) correctly identify the Delta variant with low levels of off-target probe activity across all 5 SNV sites. The 484E probe is the weakest performer, likely owing to the aforementioned mismatch in the domain region of this probe. Additionally, in some experiments, the MS2 extraction control performed poorly owing to competition for resources amongst 8 different amplicons. We also note that these runs only include 1 replicate of each the MS2 and IPC controls, rather than 2 as in Fig. 4, as this was an updated panel including the 501Y-Om2 probe. These data demonstrate the ability of our technology to accurately assess more complex samples containing multiple RNA targets of interest, showcasing the versatility of the assay.
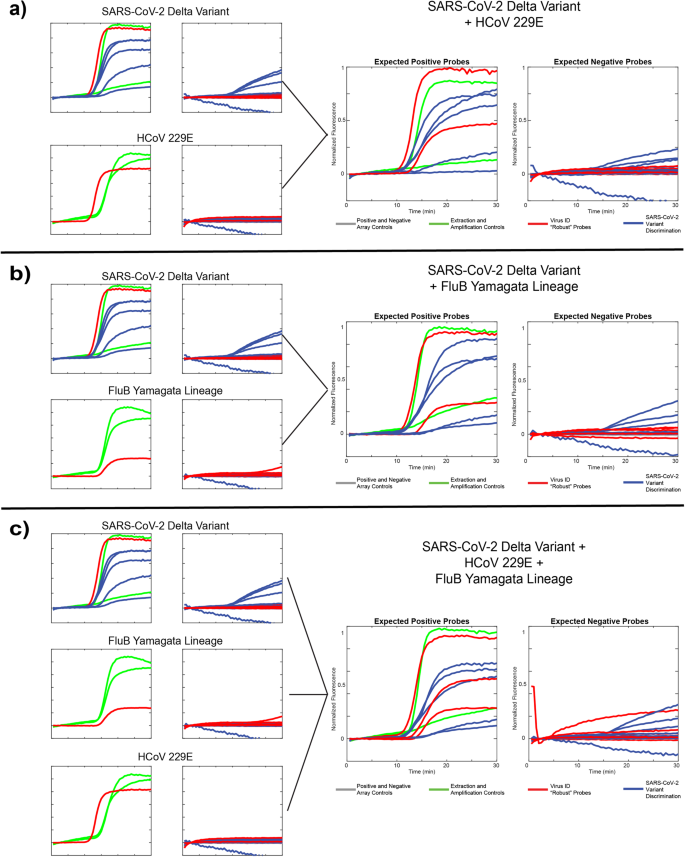
a Single-template results from the SARS-CoV-2 Delta variant and human coronavirus 229E along with a “coinfection” sample including both templates. b Single-template results from the SARS-CoV-2 Delta variant and the Yamagata lineage of influenza B, along with a coinfection sample including both templates. c. Single-target results from the same targets as in a and b, along with a coinfection sample including all three templates.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://www.nature.com/articles/s42003-023-05346-4

