
تسلط هذه المقالة الضوء على بعض أوجه التشابه والاختلاف بين المسارات التنظيمية للأجهزة الطبية 510 (ك) وعلامة CE وتساعد على تنسيق بعض جوانب الإستراتيجية التنظيمية الشاملة.
ملاحظة المحرر: تقوم هذه المقالة بتحديث واستبدال مدونة فنسنت كرابتري لعام 2014 بشأن 510 (ك) وعلامة CE (النقطة 1 والنقطة 2).
لدى إدارة الغذاء والدواء (FDA) تعريف واضح لمسار 510(ك) الذي يسمح للمصنعين بطرح منتجاتهم في السوق بوتيرة أسرع وتكلفة أقل مقارنةً بمسار الموافقة المسبقة للتسويق (PMA) الخاص بإدارة الغذاء والدواء. لم تعد عملية وضع علامة CE الخاصة بالاتحاد الأوروبي، والتي كانت تعتبر في الأصل عملية بسيطة إذا تمت الموافقة على الجهاز من قبل إدارة الغذاء والدواء، عملية تافهة. أدى تقديم MDR الخاص بالاتحاد الأوروبي (2017/745) وIVDR (2017/746) إلى قيام العديد من الشركات المصنعة ببدء برامج علاجية لتحديث وثائقهم من أجل الامتثال وإجراء اختبارات إضافية أو دراسات سريرية.
ملخص تنفيذي
تمر توجيهات الأجهزة الطبية (MDD) بمرحلة انتقالية إلى لوائح الأجهزة الطبية (MDR) في الاتحاد الأوروبي والتي تعيد تصنيف الأجهزة الطبية إلى الفئة الأولى، وIs، وIm، وIr، وIIa، وIIb، وIII، وIIIc (مخصصة). يوجه تصنيف الجهاز متطلبات التصميم والاختبار والتحقق والتحقق والفحوصات السريرية ومراقبة ما بعد السوق للجهاز الذي يحمل علامة CE ويتم طرحه في سوق الاتحاد الأوروبي.
لا يمكن مقارنة عملية 510(ك) وعملية علامة CE على أساس فردي لأن عملية 510(ك) تنطبق على عدد صغير نسبيًا من المنتجات عند مقارنتها بإمكانية تطبيق عملية علامة CE. تتطلب التغييرات المهمة في الأجهزة مستوى معينًا من المراقبة وإعداد التقارير في كلتا العمليتين وقد تؤدي إلى الحاجة إلى 510(ك) جديد أو إعادة الفحص من قبل الهيئات المبلغة.
لقد تطورت عملية 510(ك) لتصبح واضحة ومباشرة مع برنامج eSTAR ووظيفة تتبع التقدم. ال يتم حاليًا تجربة برنامج eSTAR لبوابة مشتركة بين إدارة الغذاء والدواء ووزارة الصحة الكنديةمما سيجعل العملية أكثر ملاءمة للمصنعين. إن EUDAMED قيد الإنشاء حاليًا ومن المتوقع أن يكون نظام العمل الكامل متاحًا في الربع الثالث من عام 3.
تعمل كل من عمليتي 510(ك) وعلامة CE بموجب مجموعات مختلفة من اللوائح الخاصة بنظام إدارة الجودة، ولكن يمكن استخدام ISO 13485:2016 مع التنفيذ الإضافي لبنود إدارة الأغذية والأدوية (FDA) والاتحاد الأوروبي MDR. مع اقتراح مواءمة خدمة الجودة السريعة مع ISO 13485:2016، سيصبح تنفيذ نظام إدارة الجودة أسهل بالنسبة للمصنعين. يلزم تقديم وثائق التصميم الأساسي وإدارة المخاطر، وتختلف الوثائق التكميلية حسب السوق.
المصطلحات والمعادلة والتطبيق
510(ك) عبارة عن طلب ما قبل التسويق تم تقديمه إلى إدارة الغذاء والدواء لإثبات أن الجهاز الذي سيتم تسويقه آمن وفعال، أي مكافئ إلى حد كبير (SE)، كجهاز يتم تسويقه بشكل قانوني. تحدد إدارة الغذاء والدواء الأمريكية (FDA) الجهاز (الأجهزة) التي يتم تسويقها بشكل قانوني والتي يتم رسم التكافؤ لها على أنها "المسند". الجهاز يعادل إلى حد كبير المسند إذا كان الجهاز الموضوع:
- له نفس الاستخدام المقصود مثل المسند وله نفس الخصائص التكنولوجية أو خصائص تكنولوجية مختلفة لا تثير أسئلة مختلفة تتعلق بالسلامة والفعالية.
- توضح المعلومات المقدمة إلى إدارة الغذاء والدواء (FDA) أن الجهاز آمن وفعال (أو أكثر) مثل الجهاز الذي يتم تسويقه بشكل قانوني.
انظر الشكل 1: التكافؤ الكبير بموجب إدارة الغذاء والدواء (FDA) لتقديمات 510 (ك).
تنطبق التقديمات بموجب المادة 510(ك) على أجهزة الفئة الأولى والثانية (ما لم تكن معفاة) وأجهزة الفئة الثالثة، إن أمكن.
على الرغم من أن كل دولة في الاتحاد الأوروبي، والتي يشار إليها عادةً بالدول الأعضاء، لديها سلطة مختصة مسؤولة عن الامتثال للجهاز المتعلق بتوجيهات وضع علامة CE، إلا أن مسؤولية وضع علامة CE يتم تفويضها إلى الهيئات المبلغة، لمنع تضارب المصالح، ومواءمة المتطلبات.
للحصول على علامة CE للجهاز، يتعين على الشركة المصنعة إثبات أن أجهزتها تتوافق مع متطلبات الاتحاد الأوروبي MDR. عادةً ما يتم تكليف الهيئات المبلغة بمراجعة السجل الرئيسي للجهاز والمستندات المرتبطة به للموافقة على علامة CE للجهاز. إحدى طرق إثبات الامتثال هي من خلال التكافؤ. تتطلب علامة CE، من خلال المطالبة بالتكافؤ بموجب معايير الاتحاد الأوروبي MDR، اعتبارات إضافية لإثبات التكافؤ. يتعين على الشركة المصنعة المطالبة بالتكافؤ في الخصائص التالية:
- التقنية - شروط الاستخدام والمواصفات والخصائص وطرق النشر (حيثما ينطبق ذلك) ومبادئ التشغيل ومتطلبات الأداء الحرجة
- البيولوجية - المواد أو المواد التي تتلامس مع نفس الأنسجة البشرية أو سوائل الجسم، ونوع ومدة التلامس المماثلة وخصائص إطلاق المواد (بما في ذلك منتجات التحلل والمواد القابلة للترشيح)
- السريرية – الحالة السريرية أو الغرض، وشدة المرض ومرحلته، والموقع في الجسم، والسكان، والمستخدم، والأداء النقدي في ضوء التأثير السريري المتوقع لغرض مقصود محدد.
يتطلب إثبات التكافؤ بموجب علامة CE بذل المزيد من الجهد وفي بعض الأحيان معلومات خاصة قد لا تكون متاحة لمحاولة المطالبة بالتكافؤ مع منتجات تنتمي إلى شركة مصنعة مختلفة. موافقة الهيئة المُخطرة على علامة CE غير مطلوبة للفئة الأولى بموجب معايير الاتحاد الأوروبي MDR. أجهزة الفئة الأولى معتمدة ذاتيًا بموجب معايير الاتحاد الأوروبي MDR. أجهزة الفئة الأولى التي يتم توفيرها على أنها معقمة (Is)، أو لها وظيفة قياس (Im) أو عبارة عن أداة جراحية قابلة لإعادة الاستخدام (Ir) تخضع لعلامة CE.
 وعلامة CE")
الشكل 1: التكافؤ الكبير بموجب إدارة الأغذية والعقاقير (FDA) لتقديمات 510 (ك).
التغييرات على الجهاز
قد تتطلب الأجهزة التي تخضع لتغييرات في التصميم تقديمًا جديدًا لـ 510(ك). يجب تقديم 510 (ك) جديد في الحالات التالية:
- التغييرات التي يتم إجراؤها بقصد التأثير بشكل كبير على سلامة الجهاز أو فعاليته.
- التغييرات الرئيسية في الملصقات - إضافة موانع الاستعمال، وإعادة تصنيف الأجهزة على أنها قابلة لإعادة الاستخدام من الاستخدام الفردي، وما إلى ذلك.
- التغييرات الرئيسية في التكنولوجيا والهندسة والأداء - التغييرات في آلية التحكم، ومبدأ التشغيل، وتغييرات نوع الطاقة، وما إلى ذلك.
- تغييرات المواد
- التعديلات التي تؤدي إلى تغيير كبير في ملف تعريف مخاطر الجهاز.
على الرغم من أن القائمة أعلاه تبدو واضحة، إلا أن هناك العديد من السيناريوهات التي تكون فيها الخطوط غير واضحة وقد لا يحتاج الجهاز إلى 510(ك) جديد. إرشادات إدارة الغذاء والدواء تحديد موعد إرسال 510(ك) للتغيير إلى جهاز موجود يوفر مخططات انسيابية مفصلة تساعد في اتخاذ القرار لتقديم 510 (ك) جديد. يناقش توجيه منفصل متى التغييرات في البرامج تتطلب 510 (ك) جديدة.
بموجب قانون الاتحاد الأوروبي MDR، يجب على الأجهزة التي تخضع لأي تغييرات مهمة في واحدة أو أكثر من الفئات التالية الإبلاغ عن التغيير إلى الهيئة المبلغة التي اعتمدت الجهاز.
- الغرض المقصود
- مواصفات التصميم أو الأداء
- العنصر أو المادة
- تصميم التعقيم أو التغليف الذي له تأثير على التعقيم
- نظام البرمجيات
يمكن أن يكون تعريف التغيير الكبير غامضًا. إرشادات MDCG يوفر معلومات إضافية حول التغييرات المهمة بموجب اتفاقية الاتحاد الأوروبي للأدوية المتعددة. قد تقرر الهيئة المقبولة إعادة تدقيق نظام إدارة الجودة الخاص بالشركة المصنعة أو الوثائق الفنية، حسب الحاجة.
ستشمل الإستراتيجية التنظيمية الجيدة متطلبات إدارة الغذاء والدواء (FDA) والاتحاد الأوروبي للأدوية المتعددة (MDR) لضمان استيفاءهما.
العملية والرسوم
يمكن تقديم طلبات 510 (ك) عبر الإنترنت باستخدام بوابة CDRH. تسمح بوابة CDRH للمصنعين بإرسال طلب الأجهزة الطبية باستخدام eSTAR، وهو نموذج PDF تفاعلي. توفر بوابة CDRH أيضًا أداة تتبع التقدم التي تعرض حالة التقديم. تحتوي البوابة على بعض القيود فيما يتعلق بحجم الملف وأنواعه ولكنها تسمح للمراسل بإرسال مستندات كبيرة الحجم بالبريد إلى مركز التحكم في المستندات CDRH (DCC). يمكن طرح أي جهاز تم منحه التخليص 510(ك) في السوق على الفور أثناء انتظار فحص نظام الجودة التابع لإدارة الغذاء والدواء (21 CFR 820)، والذي قد يتم بعد التخليص.
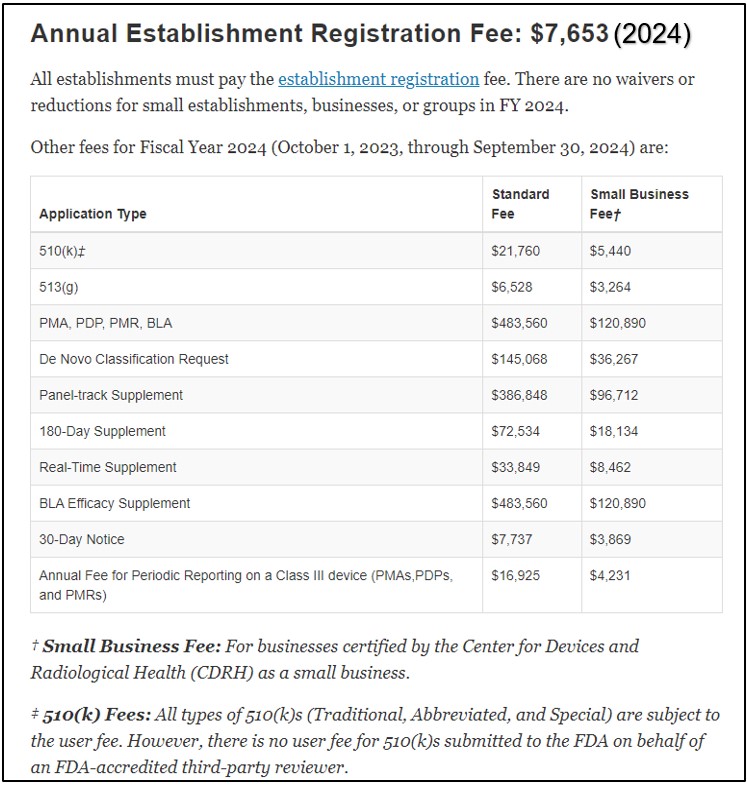
يتم نشر تكاليف طلب 510 (ك) على الموقع الإلكتروني لإدارة الغذاء والدواء (FDA) أدناه تعديلات رسوم مستخدم الأجهزة الطبية (MDUFA). تختلف الرسوم للشركات القياسية والشركات الصغيرة. يقدم الجدول أدناه فكرة عن هيكل الرسوم. يتم تحديث الرسوم كل سنة مالية. يجب على كل مؤسسة أن تدفع سنويا رسوم تسجيل المنشأة.
تتطلب عملية وضع علامة CE مشاركة الهيئات المبلغة وتدقيق مؤهل لنظام إدارة الجودة قبل أن يتم تقييم الجهاز للحصول على علامة CE. اختر الجهة المُخطرة الخاصة بك بناءً على المجموعة الصحيحة من الرسوم وترتيبات السفر والخبرة التي تناسبك.
قدمت اتفاقية الاتحاد الأوروبي للأدوية المتعددة مفهوم قاعدة البيانات الأوروبية للأجهزة الطبية (EUDAMED) التي تهدف إلى توفير "صورة حية لدورة حياة الأجهزة الطبية المتوفرة في الاتحاد الأوروبي (EU)". سيتألف EUDAMED من ست وحدات تتعلق بما يلي: تسجيل الممثل، وتحديد الجهاز الفريد (UDI) وتسجيل الجهاز، والهيئات والشهادات المبلغة، والتحقيقات السريرية ودراسات الأداء، واليقظة ومراقبة السوق. اعتبارًا من مارس 2024، أصبح لدى EUDAMED ثلاث وحدات حية - المشغلين الاقتصاديين والأجهزة والشهادات. ومن المقرر أن يتم نشر الوحدات الثلاث الأخيرة عبر الإنترنت في الربع الثالث من عام 3.
في حالة عدم وجود نظام عبر الإنترنت لإدارة الطلبات المقدمة للحصول على علامة CE، يُطلب من الشركة المصنعة استخدام البوابات/مشاركة المجلدات/الطرق الأخرى التي أعدتها الهيئات المبلغة لمشاركة الوثائق والأدلة المطلوبة للحصول على الشهادة. تجري الجهة المُبلَّغة تدقيقًا لنظام إدارة الجودة (QMS) وفقًا لمتطلبات نظام إدارة الجودة الخاص بالاتحاد الأوروبي للأدوية المتعددة (الملحق التاسع) قبل اعتماد الجهاز للسوق. لا يمكن أن تحمل الأجهزة علامة CE وطرحها في السوق دون إجراء تدقيق لنظام إدارة الجودة من هيئة مُخطرة ومراجعة الوثائق الفنية (أو أخذ عينات في بعض الحالات)، ما لم يتم تصنيف الجهاز على أنه من الفئة الأولى.
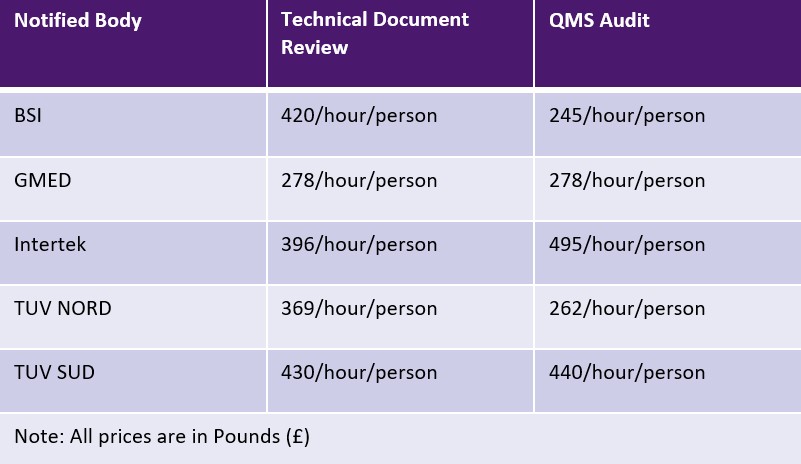
يسمح نظام EU MDR للهيئات المبلغة بفرض رسوم ثابتة أو رسوم على أساس الوقت لكل نشاط. ومع ذلك، فإن الهيئات المبلغة ليست ملزمة بأي حدود للرسوم. ويبين الجدول أدناه عينة من الجهات المبلغة ورسومها. قد يتم تطبيق رسوم إضافية للسفر والخبرة.
نظام إدارة الجودة
يُطلب من الشركات المصنعة بذل العناية الواجبة والتأكد من وجود نظام إدارة الجودة المناسب، قبل طرح أجهزتها في السوق.
تحكم إدارة الغذاء والدواء الأمريكية (FDA) نظام إدارة الجودة بموجب لوائح نظام الجودة (QSR) (21 CFR 820)، والتي تنطبق على الشركات المصنعة التي تنوي توزيع الأجهزة الطبية تجاريًا. تم إقرار قاعدة جديدة لدمج معيار ISO 13485:2016 للأجهزة الطبية – أنظمة إدارة الجودة داخل مطاعم الخدمة السريعة في محاولة لمواءمة المتطلبات في جميع المجالات. يتم استخدام ISO 13485:2016 من قبل العديد من السلطات التنظيمية الأخرى حول العالم كمعيار لأنظمة إدارة الجودة. في حين أن مرونة التخليص 510 (ك) تسمح للمصنعين بطرح أجهزتهم في السوق، إلا أنه يجوز لهيئة الغذاء والدواء فحصها في أي وقت. ومن ثم، فمن مصلحة الشركة المصنعة ضمان وجود نظام إدارة جودة متوافق قبل طرح الجهاز في السوق.
مع الاتحاد الأوروبي MDR، فإن ISO 13485:2016 ليس شرطًا لعلامة CE. يجب أن يتوافق نظام إدارة الجودة المعمول به مع اللوائح المنصوص عليها في الملحق التاسع. يجب على الشركات المصنعة إكمال طلب لتقييم نظام إدارة الجودة الخاص بهم مع الهيئة المُخطرة. بالإضافة إلى تدقيق نظام إدارة الجودة، ستقوم الهيئة المُخطرة بتقييم الوثائق الفنية للأجهزة مقابل نظام إدارة الجودة لضمان استيفاء جميع المتطلبات قبل الاعتماد.
لمواءمة نظام إدارة الجودة، فإن أفضل الممارسات هي تنفيذ نظام متوافق مع ISO 13485:2016 وبرنامج التدقيق الفردي للأجهزة الطبية (MDSAP) مع متطلبات الامتثال الإضافية لإدارة الأغذية والعقاقير (FDA) أو الاتحاد الأوروبي MDR أو وزارة الصحة الكندية أو البلدان الأخرى التي تنوي الشركة المصنعة القيام بأعمال تجارية فيها .
توثيق
تتطلب إدارة الغذاء والدواء الأمريكية (FDA) والاتحاد الأوروبي MDR عملية تصميم خاضعة للرقابة للأجهزة.
يتطلب تقديم 510(ك) ملفًا كاملاً لتاريخ التصميم (DHF) يتضمن تفاصيل متطلبات نظام الجهاز وبنيته ومواصفاته والتحقق والتحقق من صحة وتوثيق أنشطة إدارة المخاطر. يسمح ملف تعريف eSTAR 510(k) للمصنعين بإرفاق المستند ذي الصلة بكل قسم من أقسام التطبيق. توفر إدارة الغذاء والدواء الأمريكية قائمة التحقق من القبول لتوجيه الشركة المصنعة في عملية 510 (ك) والتأكد من وجود الوثائق الصحيحة.
بالنسبة لـ EU MDR، يحتوي الملف الفني على مستندات DHF ويتطلب أيضًا من الشركة المصنعة تقديم قوائم المراجعة الإضافية التالية:
- متطلبات السلامة والأداء العامة (GSPR) بموجب الملحق الأول، والمعروفة سابقًا باسم قائمة التحقق من المتطلبات الأساسية بموجب MDD
- الوثائق الفنية القياسية (SteD) بموجب الملحق الثاني
- مراقبة ما بعد السوق (PMS) بموجب الملحق الثالث
تشير الوثيقة الفنية عالية المستوى عادةً إلى مستندات DHF المختلفة وقوائم المراجعة.
وفي كلتا الحالتين، يلزم تقديم ملف إدارة مخاطر كامل متوافق مع ISO 14971.
المراجع:
- إشعار ما قبل السوق 510 (ك) | ادارة الاغذية والعقاقير
- برنامج 510 (ك): تقييم التكافؤ الكبير في إشعارات ما قبل السوق [510 (ك)] (fda.gov)
- تحديد موعد تقديم 510(ك) للتغيير إلى جهاز موجود - إرشادات لموظفي الصناعة وإدارة الغذاء والدواء (fda.gov)
- تحديد موعد تقديم 510(ك) لتغيير البرنامج إلى جهاز موجود - مسودة إرشادات لموظفي الصناعة وإدارة الغذاء والدواء (fda.gov)
- MDCG 2019-15، ملاحظات إرشادية لمصنعي الأجهزة الطبية من الفئة الأولى
- أرسل وتتبع طلبات الشراء المسبق للأجهزة الطبية عبر الإنترنت: بوابة CDRH | ادارة الاغذية والعقاقير
- قاعدة بيانات EUDAMED – EUDAMED (europa.eu)
- تعديلات رسوم مستخدم الأجهزة الطبية (MDUFA) | ادارة الاغذية والعقاقير
- MDCG 2023-2، قائمة الرسوم القياسية
- تنظيم نظام الجودة (QS)/ممارسات التصنيع الجيدة للأجهزة الطبية | ادارة الاغذية والعقاقير
- قوائم مراجعة القبول لـ 510 (ك) | ادارة الاغذية والعقاقير
- برنامج اي ستار | ادارة الاغذية والعقاقير
- FDA Health Canada eSTAR (starfishmedical.com)
الصور: أدوبي الأسهم & ستارفيش الطبية
دروفيثا كريشنا هو أخصائي ضمان الجودة / را في StarFish Medical مع درجة الماجستير في الهندسة الطبية الحيوية. عملت في التصنيع وتنفيذ المنتجات الجديدة ونشر البرامج وإدارة المشاريع والمجالات التنظيمية في شركات الأجهزة الطبية. Dhruvitha مكرس للجودة والتحسين التنظيمي والمستمر للعملية للشركة المصنعة.
- محتوى مدعوم من تحسين محركات البحث وتوزيع العلاقات العامة. تضخيم اليوم.
- PlatoData.Network Vertical Generative Ai. تمكين نفسك. الوصول هنا.
- أفلاطونايستريم. ذكاء Web3. تضخيم المعرفة. الوصول هنا.
- أفلاطون كربون، كلينتك ، الطاقة، بيئة، شمسي، إدارة المخلفات. الوصول هنا.
- أفلاطون هيلث. التكنولوجيا الحيوية وذكاء التجارب السريرية. الوصول هنا.
- المصدر https://starfishmedical.com/blog/medical-device-510k-ce-marking/

