
Bu makale, Tıbbi Cihaz 510(k) ile CE İşareti düzenleme yolları arasındaki bazı benzerlikleri ve farklılıkları vurgular ve genel bir düzenleme stratejisinin bazı yönlerinin uyumlu hale getirilmesine yardımcı olur.
Editörün Notu: Bu makale, Vincent Crabtree'nin 2014(k) ve CE İşareti (Bölüm 510 ve Bölüm 1) hakkındaki 2 blogunu günceller ve onun yerine geçer.
FDA, üreticilerin ürünlerini FDA'nın Pazarlama Öncesi Onay (PMA) yoluna kıyasla daha hızlı ve daha düşük maliyetle pazara sunmalarına olanak tanıyan 510(k) yolunun net bir tanımına sahiptir. Başlangıçta cihazın FDA onayı alması durumunda basit olarak kabul edilen Avrupa Birliği CE işaretleme süreci artık önemsiz bir süreç değil. AB'nin MDR (2017/745) ve IVDR'nin (2017/746) kullanıma sunulması, birçok üreticinin uyumluluk için belgelerini güncellemek ve ek testler veya klinik çalışmalar yürütmek üzere iyileştirme programları başlatmasına yol açtı.
Yönetici Özeti
Tıbbi Cihaz Direktifleri (MDD), tıbbi cihazları Sınıf I, Is, Im, Ir, IIa, IIb, III ve IIIc (özel) olarak yeniden sınıflandıran AB'deki Tıbbi Cihaz Yönetmeliklerine (MDR) geçiş aşamasındadır. Cihaz sınıflandırması, CE İşareti alınacak ve AB Pazarına sunulacak cihaza yönelik tasarım, test, doğrulama, validasyon, klinik ve piyasaya arz sonrası gözetim gerekliliklerini yönlendirir.
510(k) süreci ile CE İşaretleme süreci birebir olarak karşılaştırılamaz çünkü 510(k) süreci, CE İşaretleme sürecinin uygulanabilirliği ile karşılaştırıldığında nispeten az sayıda ürün için geçerlidir. Cihazlarda yapılan önemli değişiklikler, her iki süreçte de belirli düzeyde izleme ve raporlama gerektirir ve yeni bir 510(k) ihtiyacına veya Onaylanmış Kuruluşlar tarafından yeniden denetim yapılmasına yol açabilir.
510(k) süreci, eSTAR programı ve ilerleme izleme işleviyle basit olacak şekilde geliştirildi. eSTAR programının şu anda ortak bir FDA ve Health Canada portalı için pilot uygulaması yapılıyorBu da süreci üreticiler için daha uygun hale getirecek. EUDAMED şu anda yapım aşamasındadır ve tam çalışır sistemin 3 yılının 2024. çeyreğinde kullanıma sunulması beklenmektedir.
Hem 510(k) hem de CE İşaretleme süreçleri, Kalite Yönetim Sistemi için farklı düzenlemeler kapsamında çalışır ancak ISO 13485:2016, FDA ve AB MDR hükümlerinin ek uygulanmasıyla kullanılabilir. QSR'nin ISO 13485:2016 ile uyumlu hale getirilmesi önerisiyle Kalite Yönetim Sisteminin uygulanması üreticiler için daha kolay hale gelecektir. Başvuru için temel tasarım ve risk yönetimi belgeleri gereklidir ve ek belgeler pazara göre değişiklik gösterir.
Terminoloji, eşdeğerlik ve uygulama
510(k), pazarlanacak cihazın yasal olarak pazarlanan bir cihaz kadar güvenli ve etkili, yani büyük ölçüde eşdeğer (SE) olduğunu göstermek için FDA'ya yapılan bir pazarlama öncesi sunumdur. FDA, yasal olarak pazarlanan ve denkliği alınan cihazı/cihazları “yüklem” olarak tanımlar. Bir cihaz, eğer söz konusu cihaz:
- Yüklemle aynı kullanım amacına sahip olan ve aynı teknolojik özelliklere veya farklı güvenlik ve etkililik sorularını gündeme getirmeyen farklı teknolojik özelliklere sahip olan.
- FDA'ya gönderilen bilgiler, cihazın yasal olarak pazarlanan cihaz kadar (veya daha fazla) güvenli ve etkili olduğunu gösterir.
Bkz. Şekil 1: 510(k) başvuruları için FDA kapsamında Önemli Eşdeğerlik.
510(k) gönderimleri Sınıf I, II cihazlar (muaf olmadıkça) ve Sınıf III cihazlar için (varsa) geçerlidir.
Tipik olarak Üye Devletler olarak anılan Avrupa Birliği'ndeki her ülkede, CE işaretleme direktiflerine ilişkin cihaza uyumdan sorumlu bir Yetkili Otorite bulunsa da, çıkar çatışmasını önlemek için CE işaretinin sorumluluğu Onaylanmış Kuruluşlara devredilmiştir. ve gereksinimleri uyumlu hale getirin.
Cihaza CE İşareti almak için üreticinin, cihazının AB MDR gerekliliklerine uygun olduğunu göstermesi gerekir. Tipik olarak Onaylanmış Kuruluşların görevi, cihaz için CE İşaretini onaylamak üzere Cihaz Ana Kaydını ve ilgili belgeleri incelemektir. Uygunluğu göstermenin yollarından biri eşdeğerliktir. CE İşareti, AB MDR kapsamında eşdeğerlik iddiasında bulunarak, eşdeğerliği kanıtlamak için ek hususlar gerektirir. Üreticinin aşağıdaki özellikler için eşdeğerlik talebinde bulunması gerekir:
- Teknik – kullanım koşulları, spesifikasyonlar ve özellikler, dağıtım yöntemleri (varsa), çalışma ilkeleri ve kritik performans gereksinimleri
- Biyolojik - aynı insan dokuları veya vücut sıvıları ile temas halinde olan malzemeler veya maddeler, benzer tür ve temas süresi ve maddelerin salınım özellikleri (bozunma ürünleri ve sızabilenler dahil)
- Klinik – klinik durum veya amaç, hastalığın ciddiyeti ve evresi, vücuttaki bölge, popülasyon, kullanıcı, belirli bir amaç için beklenen klinik etki açısından kritik performans.
CE İşareti kapsamında eşdeğerliğin gösterilmesi, çok daha fazla çaba gerektirir ve bazen farklı bir üreticiye ait ürünlerle eşdeğerlik iddiasında bulunamayabileceğiniz özel bilgiler gerektirir. AB MDR kapsamında Sınıf I için CE İşareti için Onaylanmış Kuruluş onayı gerekli değildir. Sınıf I cihazlar AB MDR kapsamında kendi kendine sertifikalandırılmıştır. Steril olarak sunulan (Is), ölçüm fonksiyonuna sahip (Im) veya tekrar kullanılabilen cerrahi alet (Ir) olan Sınıf I cihazlar CE İşaretine tabidir.
 ve CE İşareti")
Şekil 1: 510(k) başvuruları için FDA kapsamında Önemli Eşdeğerlik.
Cihazdaki Değişiklikler
Tasarım değişikliğine uğrayan cihazlar, yeni bir 510(k) gönderimi gerektirebilir. Aşağıdaki durumlarda yeni bir 510(k) sunulmalıdır:
- Bir cihazın güvenliğini veya etkinliğini önemli ölçüde etkilemek amacıyla yapılan değişiklikler.
- Önemli Etiketleme değişiklikleri – Kontrendikasyonların eklenmesi, cihazların tek kullanımdan itibaren yeniden kullanılabilir olarak yeniden etiketlenmesi vb.
- Büyük Teknoloji, mühendislik ve performans değişiklikleri – kontrol mekanizmasındaki değişiklikler, çalışma prensibi, enerji türü değişiklikleri vb.
- Malzeme değişiklikleri
- Cihazın risk profilinde önemli değişikliğe yol açan değişiklikler.
Yukarıdaki liste basit gibi görünse de çizgilerin bulanıklaştığı ve cihazın yeni bir 510(k) gerektirmediği çeşitli senaryolar vardır. FDA rehberliği Mevcut Bir Cihazda Değişiklik İçin 510(k)'nın Ne Zaman Gönderileceğine Karar Verme yeni bir 510(k) sunulmasına yönelik karar vermeye yardımcı olan ayrıntılı akış şemaları sağlar. Ayrı bir kılavuzda ne zaman yazılımdaki değişiklikler yeni bir 510(k) gerektirir.
AB MDR uyarınca, aşağıdaki kategorilerden bir veya daha fazlasında önemli değişikliklere uğrayan cihazların, değişikliği, cihazı onaylayan Onaylanmış Kuruluşa bildirmesi gerekir.
- kullanım amacı
- tasarım veya performans spesifikasyonu
- bileşen veya malzeme
- Sterilizasyona etkisi olan sterilizasyon veya ambalaj tasarımı
- yazılım
Önemli değişimin tanımı belirsiz olabilir. MDCG Rehberi AB MDR kapsamındaki önemli değişiklikler hakkında ek bilgi sağlar. Onaylanmış Kuruluş, gerektiğinde üreticinin KYS'sini veya teknik dokümantasyonunu yeniden denetlemeye karar verebilir.
İyi bir düzenleme stratejisi, her ikisinin de karşılandığından emin olmak için hem FDA hem de AB MDR gerekliliklerinden oluşacaktır.
Süreç ve Ücretler
510(k) başvuruları CDRH portalı kullanılarak çevrimiçi olarak yapılabilir. CDRH portalı, üreticilerin etkileşimli bir PDF formu olan eSTAR'ı kullanarak Tıbbi Cihaz gönderimi göndermesine olanak tanır. CDRH portalı ayrıca gönderim durumunu görüntüleyen bir ilerleme izleyicisi de sağlar. Portalın dosya boyutu ve türleriyle ilgili birkaç sınırlaması vardır ancak muhabirin büyük boyutlu belgeleri CDRH Belge Kontrol Merkezine (DCC) postayla göndermesine olanak tanır. 510(k) izni verilen herhangi bir cihaz, iznin ardından gerçekleşebilecek olan FDA Kalite Sistemi (21 CFR 820) incelemesini beklerken hemen piyasaya sürülebilir.
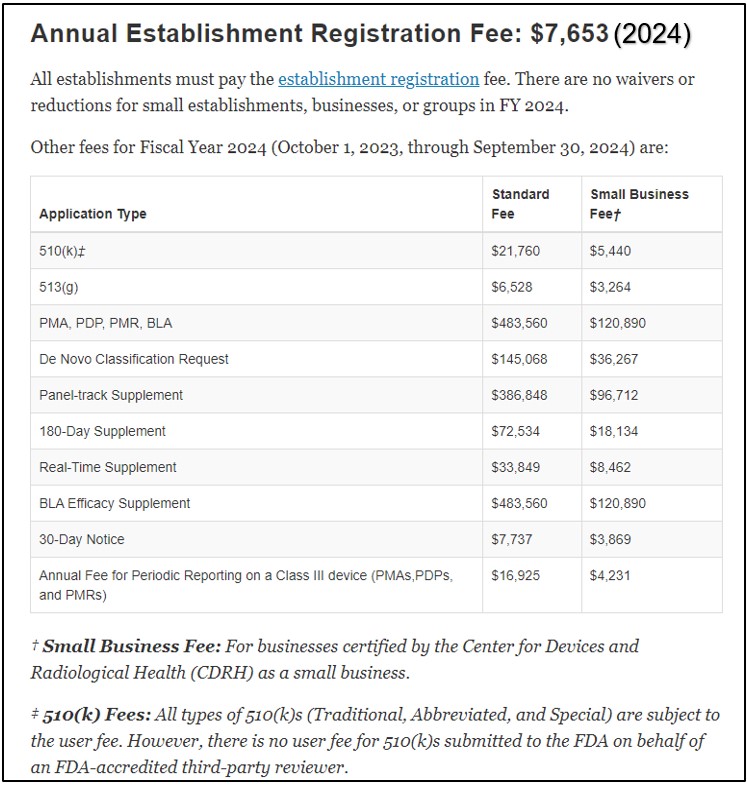
510(k) için başvuru maliyetleri FDA web sitesinde şu şekilde yayınlanmaktadır: Tıbbi Cihaz Kullanıcı Ücreti Değişiklikleri (MDUFA). Ücretler standart işletmeler ve küçük işletmeler için farklılık gösterir. Aşağıdaki tablo ücret yapısına ilişkin bir fikir vermektedir. Ücretler her mali yılda güncellenir. Her kuruluşun yıllık ücret ödemesi gerekmektedir. kuruluş kayıt ücreti.
CE İşaretleme süreci, bir cihazın CE İşareti için değerlendirilmesinden önce Onaylanmış Kuruluşların katılımını ve nitelikli bir KYS Denetimi yapılmasını gerektirir. Sizin için uygun olan doğru ücret, seyahat düzenlemeleri ve uzmanlık kombinasyonuna göre Onaylanmış Kuruluşunuzu seçin.
AB MDR, "Avrupa Birliği'nde (AB) kullanıma sunulan tıbbi cihazların yaşam döngüsünün canlı bir resmini" sağlamayı amaçlayan Avrupa Tıbbi Cihazlar Veri Tabanı (EUDAMED) veri tabanı kavramını tanıttı. EUDAMED, aktör kaydı, benzersiz cihaz tanımlama (UDI) ve cihaz kaydı, Onaylanmış Kuruluşlar ve sertifikalar, klinik araştırmalar ve performans çalışmaları, vijilans ve piyasa gözetimi ile ilgili altı modülden oluşacaktır. Mart 2024 itibarıyla EUDAMED'in ekonomik operatörler, cihazlar ve sertifikalar olmak üzere üç canlı modülü bulunmaktadır. Son üç modülün 3 yılının 2024. çeyreğinde çevrimiçi olması planlanıyor.
CE İşareti başvurularını yönetecek çevrimiçi bir sistemin bulunmaması durumunda, üreticinin sertifikasyon için gerekli belge ve kanıtları paylaşmak üzere Onaylanmış Kuruluşlar tarafından oluşturulan portalları/dosya klasörlerini/diğer yöntemleri kullanması gerekir. Onaylanmış Kuruluş, cihazı pazar için sertifikalandırmadan önce AB MDR Kalite Yönetim Sistemi gerekliliklerine (Ek IX) göre bir KYS Denetimi gerçekleştirir. Cihazlar, Sınıf I olarak sınıflandırılmadığı sürece, Onaylanmış Kuruluş KYS denetimi ve teknik dokümantasyon incelemesi (veya bazı durumlarda örnekleme) olmadan CE işareti alamaz ve piyasaya sürülemez.
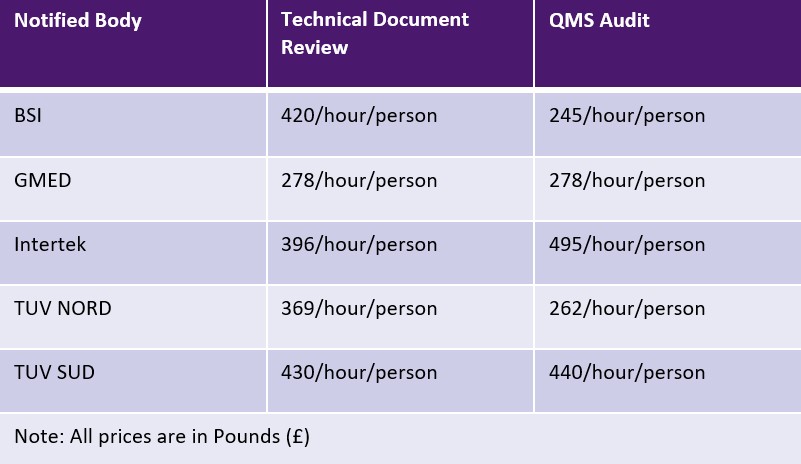
AB MDR, Onaylanmış Kuruluşların her faaliyet için sabit bir ücret veya zamana dayalı bir ücret talep etmesine olanak tanır. Ancak Onaylanmış Kuruluşlar ücretlere ilişkin herhangi bir sınırlamaya tabi değildir. Aşağıdaki tablo Onaylanmış Kuruluşların örneklerini ve ücretlerini göstermektedir. Seyahat ve uzmanlık için ek ücretler geçerli olabilir.
Kalite Yönetim Sistemi
Üreticilerin, cihazlarını piyasaya sürmeden önce gerekli özeni göstermeleri ve uygun Kalite Yönetim Sisteminin yürürlükte olduğundan emin olmaları gerekmektedir.
FDA, tıbbi cihazları ticari olarak dağıtmayı amaçlayan üreticiler için geçerli olan Kalite Sistem Düzenlemeleri (QSR) (21 CFR 820) kapsamında kalite yönetim sistemini yönetir. Gereklilikleri tüm alanda uyumlu hale getirmek amacıyla ISO 13485:2016 Tıbbi Cihazlar – Kalite Yönetim Sistemlerini QSR'ye dahil etmek için yeni bir kural kabul edildi. ISO 13485:2016, dünya çapındaki diğer birçok düzenleyici otorite tarafından Kalite Yönetim Sistemleri standardı olarak kullanılmaktadır. 510(k) izninin esnekliği, üreticilerin cihazlarını piyasaya sürmesine olanak tanırken, istedikleri zaman FDA tarafından denetlenebilirler. Bu nedenle, cihazı piyasaya sürmeden önce uyumlu bir Kalite Yönetim Sisteminin mevcut olduğundan emin olmak üreticinin çıkarınadır.
EU MDR ile ISO 13485:2016, CE İşareti için bir gereklilik değildir. Uygulanan Kalite Yönetim Sistemi Ek IX'da belirtilen düzenlemelere uygun olmalıdır. Üreticiler, Kalite Yönetim Sisteminin değerlendirilmesi için Onaylanmış Kuruluşa başvuruda bulunmalıdır. Onaylanmış Kuruluş, Kalite Yönetim Sistemi denetimine ek olarak, belgelendirmeden önce tüm gerekliliklerin karşılandığından emin olmak için cihazların teknik dokümantasyonunu Kalite Yönetim Sistemine göre değerlendirecektir.
Kalite Yönetim Sistemini uyumlu hale getirmek için en iyi uygulama, ISO 13485:2016 ve Tıbbi Cihaz Tek Denetim Programı (MDSAP) uyumlu bir sistemin, FDA, AB MDR, Kanada Sağlık Bakanlığı veya üreticinin iş yapmayı planladığı diğer ülkeler için ek uyumluluk gereksinimleriyle birlikte uygulanmasıdır. .
belgeleme
FDA ve EU MDR, cihazlar için kontrollü bir tasarım süreci gerektirir.
510(k) gönderimi, cihaz sistem gereksinimlerini, mimarisini, spesifikasyonlarını, doğrulanmasını ve onaylanmasını ve risk yönetimi faaliyetlerinin belgelenmesini detaylandıran eksiksiz bir Tasarım Geçmişi Dosyası (DHF) gerektirir. 510(k) eSTAR profili, üreticilerin ilgili belgeyi başvurunun her bölümüne eklemesine olanak tanır. FDA şunları sağlar: Kabul Kontrol Listesi Üreticiye 510(k) süreci konusunda rehberlik etmek ve doğru belgelerin mevcut olduğundan emin olmak.
AB MDR için bir Teknik Dosya, DHF belgelerini içerir ve ayrıca üreticinin aşağıdaki ek kontrol listelerini sunmasını gerektirir:
- Daha önce MDD kapsamında Temel Gereksinimler Kontrol Listesi olarak bilinen Ek I kapsamındaki Genel Güvenlik ve Performans Gereksinimleri (GSPR)
- Ek II kapsamında Standart Teknik Dokümantasyon (SteD)
- Ek III kapsamında piyasaya arz sonrası gözetim (PMS)
Üst düzey bir teknik belge genellikle çeşitli DHF belgelerine ve kontrol listelerine atıfta bulunur.
Her iki durumda da ISO 14971 ile uyumlu tam bir risk yönetimi dosyası gereklidir.
Referanslar:
- Piyasa Öncesi Bildirim 510(k) | FDA
- 510(k) Programı: Piyasa Öncesi Bildirimlerde Önemli Eşdeğerliğin Değerlendirilmesi [510(k)] (fda.gov)
- Mevcut Bir Cihazda Değişiklik İçin 510(k)'nın Ne Zaman Gönderileceğine Karar Verme - Endüstri ve Gıda ve İlaç İdaresi Personeli için Kılavuz (fda.gov)
- Mevcut Bir Cihazda Yazılım Değişikliği İçin 510(k)'nin Ne Zaman Gönderileceğine Karar Verme - Endüstri ve Gıda ve İlaç İdaresi Personeli için Taslak Kılavuz (fda.gov)
- MDCG 2019-15, Sınıf I Tıbbi Cihaz Üreticileri için Kılavuz Notları
- Tıbbi Cihaz Pazar Öncesi Başvurularını Çevrimiçi Gönderin ve Takip Edin: CDRH Portalı | FDA
- EUDAMED veritabanı – EUDAMED (europa.eu)
- Tıbbi Cihaz Kullanıcı Ücreti Değişiklikleri (MDUFA) | FDA
- MDCG 2023-2, Standart Ücretler Listesi
- Kalite Sistemi (QS) Yönetmeliği/Tıbbi Cihaz İyi Üretim Uygulamaları | FDA
- 510(k)'ler için Kabul Kontrol Listeleri | FDA
- eSTAR Programı | FDA
- FDA Sağlık Kanada eSTAR (starfishmedical.com)
resimler: Adobe Stock & StarFish Medikal
Dhruvitha Krishna bir QA/RA Uzmanı StarFish Medical'da Biyomedikal Mühendisliği alanında yüksek lisans derecesine sahip. Tıbbi cihaz şirketlerinde üretim, yeni ürün uygulaması, yazılım dağıtımı, proje yönetimi ve mevzuat alanlarında çalıştı. Dhruvitha kendisini üretici için kaliteye, mevzuata ve sürekli süreç iyileştirmeye adamıştır.
- SEO Destekli İçerik ve Halkla İlişkiler Dağıtımı. Bugün Gücünüzü Artırın.
- PlatoData.Network Dikey Üretken Yapay Zeka. Kendine güç ver. Buradan Erişin.
- PlatoAiStream. Web3 Zekası. Bilgi Genişletildi. Buradan Erişin.
- PlatoESG. karbon, temiz teknoloji, Enerji, Çevre, Güneş, Atık Yönetimi. Buradan Erişin.
- PlatoSağlık. Biyoteknoloji ve Klinik Araştırmalar Zekası. Buradan Erişin.
- Kaynak: https://starfishmedical.com/blog/medical-device-510k-ce-marking/

