Eiwitten sturen veel biologische processen aan, zoals enzymactiviteit, moleculair transport en cellulaire ondersteuning. De driedimensionale structuur van een eiwit geeft inzicht in zijn functie en hoe het interageert met andere biomoleculen. Experimentele methoden om de eiwitstructuur te bepalen, zoals röntgenkristallografie en NMR-spectroscopie, zijn duur en tijdrovend.
Daarentegen kunnen recent ontwikkelde computationele methoden snel en nauwkeurig de structuur van een eiwit voorspellen op basis van de aminozuursequentie. Deze methoden zijn van cruciaal belang voor eiwitten die moeilijk experimenteel te bestuderen zijn, zoals membraaneiwitten, de doelwitten van veel geneesmiddelen. Een bekend voorbeeld hiervan is AlphaFold, een op deep learning gebaseerd algoritme dat bekend staat om zijn nauwkeurige voorspellingen.
ESMFold is een andere zeer nauwkeurige, op diep leren gebaseerde methode die is ontwikkeld om de eiwitstructuur te voorspellen op basis van de aminozuursequentie. ESMFold gebruikt een groot eiwittaalmodel (pLM) als ruggengraat en werkt end-to-end. In tegenstelling tot AlphaFold2 hoeft het niet te worden opgezocht of Uitlijning van meerdere reeksen (MSA) stap, en het is ook niet afhankelijk van externe databases om voorspellingen te genereren. In plaats daarvan heeft het ontwikkelingsteam het model getraind op miljoenen eiwitsequenties van UniRef. Tijdens de training ontwikkelde het model aandachtspatronen die op elegante wijze de evolutionaire interacties tussen aminozuren in de reeks weergeven. Dit gebruik van een pLM in plaats van een MSA maakt tot 60 keer snellere voorspellingstijden mogelijk dan andere geavanceerde modellen.
In deze post gebruiken we het vooraf getrainde ESMFold-model van Hugging Face with Amazon Sage Maker om de zware ketenstructuur van te voorspellen trastuzumab, een monoklonaal antilichaam voor het eerst ontwikkeld door Genentech voor de behandeling van HER2-positieve borstkanker. Het snel voorspellen van de structuur van dit eiwit zou handig kunnen zijn als onderzoekers het effect van sequentiemodificaties willen testen. Dit kan mogelijk leiden tot een betere overleving van de patiënt of minder bijwerkingen.
Dit bericht bevat een voorbeeld van een Jupyter-notebook en gerelateerde scripts in het volgende GitHub-repository.
Voorwaarden
We raden aan dit voorbeeld uit te voeren in een Amazon SageMaker Studio notitieboekje het uitvoeren van de PyTorch 1.13 Python 3.9 CPU-geoptimaliseerde afbeelding op een ml.r5.xlarge instantietype.
Visualiseer de experimentele structuur van trastuzumab
Om te beginnen gebruiken we de biopython bibliotheek en een helperscript om de trastuzumab-structuur te downloaden van de RCSB Eiwitgegevensbank:
Vervolgens gebruiken we de py3Dmol bibliotheek om de structuur te visualiseren als een interactieve 3D-visualisatie:
De volgende afbeelding geeft de 3D-eiwitstructuur 1N8Z uit de Protein Data Bank (PDB) weer. In deze afbeelding wordt de lichte keten van trastuzumab oranje weergegeven, is de zware keten blauw (met het variabele gebied in lichtblauw) en is het HER2-antigeen groen.

We zullen eerst ESMFold gebruiken om de structuur van de zware keten (keten B) te voorspellen op basis van de aminozuursequentie. Vervolgens zullen we de voorspelling vergelijken met de hierboven getoonde experimenteel bepaalde structuur.
Voorspel de structuur van de zware keten van trastuzumab op basis van de volgorde met behulp van ESMFold
Laten we het ESMFold-model gebruiken om de structuur van de zware keten te voorspellen en deze te vergelijken met het experimentele resultaat. Om te beginnen gebruiken we een vooraf gebouwde notebookomgeving in Studio die wordt geleverd met verschillende belangrijke bibliotheken, zoals PyTorch, vooraf geïnstalleerd. Hoewel we een versneld instantietype zouden kunnen gebruiken om de prestaties van onze notebookanalyse te verbeteren, gebruiken we in plaats daarvan een niet-versnelde instantie en voeren we de ESMFold-voorspelling uit op een CPU.
Eerst laden we het vooraf getrainde ESMFold-model en de tokenizer van Knuffelen Gezicht Hub:
Vervolgens kopiëren we het model naar ons apparaat (in dit geval CPU) en stellen we enkele modelparameters in:
Om de eiwitsequentie voor analyse voor te bereiden, moeten we deze tokeniseren. Dit vertaalt de aminozuursymbolen (EVQLV…) in een numeriek formaat dat het ESMFold-model kan begrijpen (6,19,5,10,19,…):
Vervolgens kopiëren we de getokeniseerde invoer naar de modus, doen we een voorspelling en slaan we het resultaat op in een bestand:
Dit duurt ongeveer 3 minuten op een niet-versneld instantietype, zoals een r5.
We kunnen de nauwkeurigheid van de ESMfold-voorspelling controleren door deze te vergelijken met de experimentele structuur. Dit doen we aan de hand van de US-uitlijnen tool ontwikkeld door het Zhang Lab aan de Universiteit van Michigan:
| PDBketen1 | PDBketen2 | TM-score |
| data/voorspelling.pdb:A | data/experimenteel.pdb:B | 0.802 |
De sjabloonmodelleringsscore (TM-score) is een maatstaf voor het beoordelen van de gelijkenis van eiwitstructuren. Een score van 1.0 duidt op een perfecte match. Scores boven de 0.7 geven aan dat eiwitten dezelfde ruggengraatstructuur delen. Scores boven de 0.9 geven aan dat het de eiwitten zijn functioneel uitwisselbaar voor stroomafwaarts gebruik. In ons geval van het behalen van TM-Score 0.802, zou de ESMFold-voorspelling waarschijnlijk geschikt zijn voor toepassingen zoals structuurscoring of ligandbindingsexperimenten, maar mogelijk niet geschikt voor gebruikssituaties zoals moleculaire vervanging die een extreem hoge nauwkeurigheid vereisen.
We kunnen dit resultaat valideren door de uitgelijnde structuren te visualiseren. De twee structuren vertonen een hoge, maar niet perfecte mate van overlap. Eiwitstructuurvoorspellingen zijn een snel evoluerend veld en veel onderzoeksteams ontwikkelen steeds nauwkeurigere algoritmen!
Implementeer ESMFold als een SageMaker-inferentie-eindpunt
Het uitvoeren van modelinferentie in een notebook is prima om mee te experimenteren, maar wat als u uw model moet integreren met een toepassing? Of een MLOps-pijplijn? In dit geval is het een betere optie om uw model te implementeren als een inferentie-eindpunt. In het volgende voorbeeld implementeren we ESMFold als een real-time deductie-eindpunt van SageMaker op een versneld exemplaar. SageMaker real-time endpoints bieden een schaalbare, kosteneffectieve en veilige manier om machine learning (ML)-modellen te implementeren en te hosten. Met automatische schaling kunt u het aantal instances waarop het endpoint draait aanpassen aan de eisen van uw applicatie, waardoor de kosten worden geoptimaliseerd en een hoge beschikbaarheid wordt gegarandeerd.
De voorgebouwde SageMaker-container voor Hugging Face maakt het gemakkelijk om deep learning-modellen in te zetten voor veelvoorkomende taken. Voor nieuwe use-cases, zoals voorspelling van de eiwitstructuur, moeten we echter een gewoonte definiëren inference.py script om het model te laden, de voorspelling uit te voeren en de uitvoer op te maken. Dit script bevat veel van dezelfde code die we in ons notitieblok gebruikten. We maken ook een requirements.txt bestand om enkele Python-afhankelijkheden te definiëren die ons eindpunt kan gebruiken. U kunt de bestanden zien die we hebben gemaakt in de GitHub-repository.
In de volgende afbeelding lijken de experimentele (blauwe) en voorspelde (rode) structuren van de zware keten van trastuzumab sterk op elkaar, maar zijn ze niet identiek.

Nadat we de benodigde bestanden in het code directory, implementeren we ons model met behulp van de SageMaker HuggingFaceModel klas. Dit maakt gebruik van een vooraf gebouwde container om het proces van het implementeren van Hugging Face-modellen naar SageMaker te vereenvoudigen. Houd er rekening mee dat het 10 minuten of langer kan duren om het eindpunt te maken, afhankelijk van de beschikbaarheid van ml.g4dn instantietypen in onze regio.
Wanneer de implementatie van het eindpunt is voltooid, kunnen we de eiwitsequentie opnieuw indienen en de eerste paar rijen van de voorspelling weergeven:
Omdat we ons eindpunt in een versnelde instantie hebben geïmplementeerd, duurt de voorspelling slechts enkele seconden. Elke rij in het resultaat komt overeen met een enkel atoom en omvat de aminozuuridentiteit, drie ruimtelijke coördinaten en een pLDDT-score die het voorspellingsvertrouwen op die locatie weergeeft.
| VOB_GROUP | ID | ATOM_LABEL | RES_ID | CHAIN_ID | SEQ_ID | CARTN_X | CARTN_Y | CARTN_Z | BEZETTING | PLDDT | ATOM_ID |
| ATOM | 1 | N | GLU | A | 1 | 14.578 | -19.953 | 1.47 | 1 | 0.83 | N |
| ATOM | 2 | CA | GLU | A | 1 | 13.166 | -19.595 | 1.577 | 1 | 0.84 | C |
| ATOM | 3 | CA | GLU | A | 1 | 12.737 | -18.693 | 0.423 | 1 | 0.86 | C |
| ATOM | 4 | CB | GLU | A | 1 | 12.886 | -18.906 | 2.915 | 1 | 0.8 | C |
| ATOM | 5 | O | GLU | A | 1 | 13.417 | -17.715 | 0.106 | 1 | 0.83 | O |
| ATOM | 6 | cg | GLU | A | 1 | 11.407 | -18.694 | 3.2 | 1 | 0.71 | C |
| ATOM | 7 | cd | GLU | A | 1 | 11.141 | -18.042 | 4.548 | 1 | 0.68 | C |
| ATOM | 8 | OE1 | GLU | A | 1 | 12.108 | -17.805 | 5.307 | 1 | 0.68 | O |
| ATOM | 9 | OE2 | GLU | A | 1 | 9.958 | -17.767 | 4.847 | 1 | 0.61 | O |
| ATOM | 10 | N | VAL | A | 2 | 11.678 | -19.063 | -0.258 | 1 | 0.87 | N |
| ATOM | 11 | CA | VAL | A | 2 | 11.207 | -18.309 | -1.415 | 1 | 0.87 | C |
Door dezelfde methode te gebruiken als voorheen, zien we dat de voorspellingen van de notebook en het eindpunt identiek zijn.
| PDBketen1 | PDBketen2 | TM-score |
| data/endpoint_prediction.pdb:A | data/voorspelling.pdb:A | 1.0 |
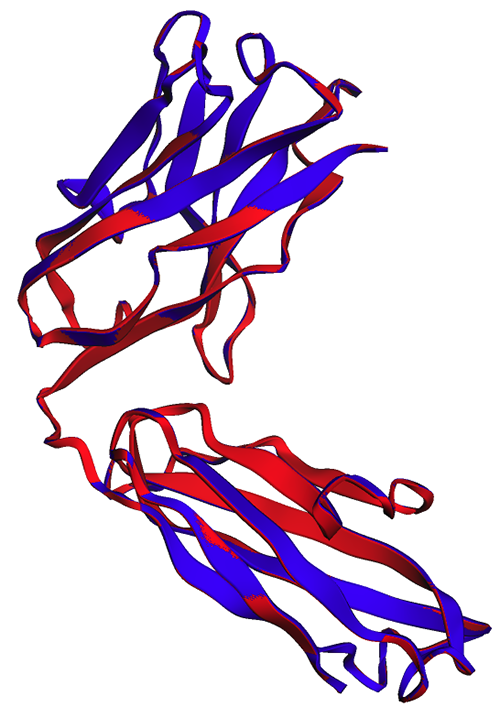
Zoals te zien is in de volgende afbeelding, laten de ESMFold-voorspellingen die in de notebook (rood) en door het eindpunt (blauw) zijn gegenereerd, een perfecte uitlijning zien.
Opruimen
Om verdere kosten te voorkomen, verwijderen we ons inferentie-eindpunt en testgegevens:
Samengevat
Computationele eiwitstructuurvoorspelling is een cruciaal hulpmiddel om de functie van eiwitten te begrijpen. Naast fundamenteel onderzoek hebben algoritmen zoals AlphaFold en ESMFold veel toepassingen in de geneeskunde en biotechnologie. De structurele inzichten die door deze modellen worden gegenereerd, helpen ons beter te begrijpen hoe biomoleculen op elkaar inwerken. Dit kan vervolgens leiden tot betere diagnostische hulpmiddelen en therapieën voor patiënten.
In dit bericht laten we zien hoe het ESMFold-eiwittaalmodel van Hugging Face Hub kan worden ingezet als een schaalbaar inferentie-eindpunt met behulp van SageMaker. Raadpleeg voor meer informatie over het implementeren van Hugging Face-modellen op SageMaker Hugging Face gebruiken met Amazon SageMaker. Je kunt ook meer voorbeelden van eiwitwetenschap vinden in de Geweldige eiwitanalyse op AWS GitHub-opslagplaats. Laat een reactie achter als er nog andere voorbeelden zijn die je graag zou willen zien!
Over de auteurs
 Brian trouw is een Senior AI/ML Solutions Architect in het Global Healthcare and Life Sciences-team bij Amazon Web Services. Hij heeft meer dan 17 jaar ervaring in biotechnologie en machine learning en is gepassioneerd om klanten te helpen bij het oplossen van genomische en proteomische uitdagingen. In zijn vrije tijd kookt en eet hij graag met zijn vrienden en familie.
Brian trouw is een Senior AI/ML Solutions Architect in het Global Healthcare and Life Sciences-team bij Amazon Web Services. Hij heeft meer dan 17 jaar ervaring in biotechnologie en machine learning en is gepassioneerd om klanten te helpen bij het oplossen van genomische en proteomische uitdagingen. In zijn vrije tijd kookt en eet hij graag met zijn vrienden en familie.
 Shamika Ariyawansa is een AI/ML Specialist Solutions Architect in het Global Healthcare and Life Sciences-team bij Amazon Web Services. Hij werkt gepassioneerd samen met klanten om hun AI- en ML-adoptie te versnellen door technische begeleiding te bieden en hen te helpen bij het innoveren en bouwen van veilige cloudoplossingen op AWS. Buiten zijn werk houdt hij van skiën en off-road rijden.
Shamika Ariyawansa is een AI/ML Specialist Solutions Architect in het Global Healthcare and Life Sciences-team bij Amazon Web Services. Hij werkt gepassioneerd samen met klanten om hun AI- en ML-adoptie te versnellen door technische begeleiding te bieden en hen te helpen bij het innoveren en bouwen van veilige cloudoplossingen op AWS. Buiten zijn werk houdt hij van skiën en off-road rijden.
 Yanjun Qi is Senior Applied Science Manager bij het AWS Machine Learning Solution Lab. Ze innoveert en past machine learning toe om AWS-klanten te helpen hun AI- en cloudadoptie te versnellen.
Yanjun Qi is Senior Applied Science Manager bij het AWS Machine Learning Solution Lab. Ze innoveert en past machine learning toe om AWS-klanten te helpen hun AI- en cloudadoptie te versnellen.
- Door SEO aangedreven content en PR-distributie. Word vandaag nog versterkt.
- PlatoAiStream. Web3 gegevensintelligentie. Kennis versterkt. Toegang hier.
- De toekomst slaan met Adryenn Ashley. Toegang hier.
- Koop en verkoop aandelen in PRE-IPO-bedrijven met PREIPO®. Toegang hier.
- Bron: https://aws.amazon.com/blogs/machine-learning/accelerate-protein-structure-prediction-with-the-esmfold-language-model-on-amazon-sagemaker/

