コンピューテーショナルデザイン戦略
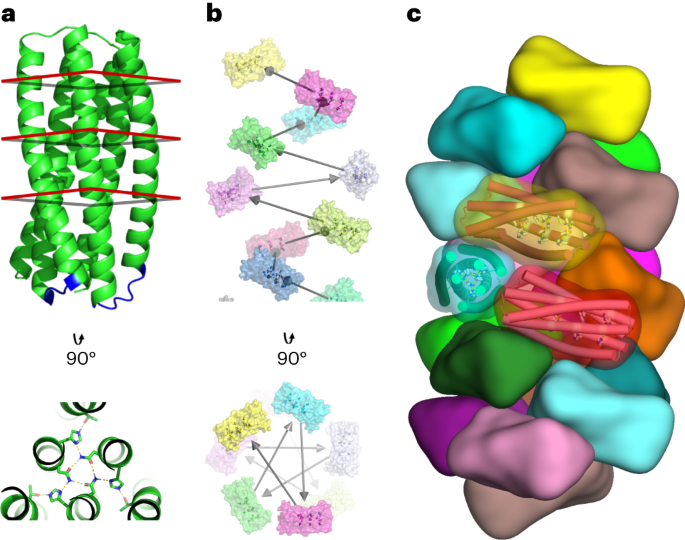
pRO-2.3 ヘリックスを XNUMX 本の鎖に接続するための短いループは、DSSP によって高解像度の結晶構造で同定された XNUMX つのヘリックス領域にまたがるフラグメントで構成されるバックボーン サンプルの網羅的なデータベースを使用して設計されました(前述のとおり)14)。ループは、最適化された重ね合わせアルゴリズムを使用したフラグメントとターゲットの末端残基の厳密なアラインメントによってこのデータベースで特定されました。15。 0.35 Å RMSD のアライメント許容値を満たす候補は、ねじれ空間座標と、アライメントされた候補バックボーンの重原子座標に対するソフト座標制約を介してターゲット バックボーンにアライメントされました。次いで、ソース構造データベースに対するループバックボーンのアラインメントを介して生成された配列プロファイル制約の下で、候補ループ配列を設計した。スコアが最も低い候補が最終的なループ設計に選択されました。
ヘリカルドッキングと設計方法7 をリンクされた pRO-2.3 に適用して、らせん状フィラメント設計モデルを生成しました。次の基準で個々の設計軌跡をフィルタリングしました: 結合 (ポリマー) 状態と非結合 (モノマー) 状態の間の -15.0 ロゼッタ エネルギー単位を超える不一致、700 Å を超える界面表面積2、ロゼッタ形状の相補性が 0.62 を超え、不満足な極性残基数が 5 未満であること。これらの基準を満たす設計は、界面の結合状態の安定化に寄与していないとみなされる突然変異への単一点復帰を含む手動の改良を受けました。各ドッキング構成の最高スコアのデザインは、実験検証のために最終的なタンパク質セットに統合されました。
タンパク質の発現と精製
合計 18 のデザインの合成遺伝子は、 大腸菌 IDT から取得し、pET29b+ ベクターの NdeI 制限部位と XhoI 制限部位の間のマルチ クローニング サイトに挿入します。これらのコンストラクトは BL21* (DE3) に導入されました E. 大腸菌の コンピテントセル。形質転換体を、50 mg l を補充した 200 ml のテリフィック ブロス培地で培養しました。-1 カナマイシン。 T7 プロモーターの制御下で、Studier 自己誘導を使用して発現を 24 °C で 37 時間進行させました。16 培養物が遠心分離によって収集されるまで。細胞ペレットをトリス緩衝生理食塩水 (TBS) に再懸濁し、Bugbuster 洗剤で溶解しました。遠心分離により清澄化された可溶性画分は、Ni による精製を受けました。2+ Ni-NTA Superflow 樹脂を使用した固定化金属アフィニティークロマトグラフィー。細胞溶解物が結合した樹脂を、40カラム容量の500mMイミダゾールおよび400mM NaClで洗浄し、続いて75mMイミダゾールおよび0.5mM NaClで溶出した。可溶性画分と不溶性画分を SDS ポリアクリルアミドゲル電気泳動分析に供しました。正しい分子量でタンパク質バンドを示すサンプルが、電子顕微鏡スクリーニング用に選択されました。選択したデザインをさらなる特性評価のために 24 μl までスケールアップし、Studier 自己誘導を使用して 37 °C で XNUMX 時間発現を再度進めました。16 遠心分離による回収前。細胞ペレットをTBSに再懸濁し、マイクロ流動化によって溶解し、続いて上記のように精製した。
ネガティブステインEM
電子顕微鏡スクリーニングのために、可溶性画分をTBS (25 mM Tris緩衝液、75 mM NaCl、pH 8)中で濃縮した。 6 µl の液滴(1 µl のサンプルを 5 µl のバッファーで瞬時に希釈)を、負グロー放電したカーボンコーティングされた 200 メッシュの銅グリッド上に塗布し、Milli-Q 水で洗浄し、0.75% ギ酸ウラニル (pH 4.0) のいずれかを使用して染色しました。 ) または前述のように Nanoprobes, Inc. から購入した Nano-W (pH 6.8)17。スクリーニングは、100 kV Morgagni M268 透過型電子顕微鏡 (FEI) または 120 kV Talos L120C 透過型電子顕微鏡 (ThermoFisher) のいずれかを使用して実施されました。画像はボトムマウントの Teitz CMOS 4k カメラ システムを使用してキャプチャされ、フィジー ソフトウェア (バージョン: 2.14.0/1.54f) を使用してコントラストが強化されるように処理されました。18 明確にするために。
ファイバーの長さは、cryoSPARC のファイバー トレース アルゴリズムを使用して定量化されました。8。この方法では、テンプレート クラスとの相互相関によって繊維を識別し、識別された粒子から連続した繊維を追跡します。 DpHF19 から生成されたテンプレート クラスが、測定されたすべてのファイバーに使用されました。繊維は平均曲率 (<0.0005 Å) に従ってフィルタリングされました。-1) と各ファイバーにわたる平均の正規化された相互相関 (>0.5)。 DpHF18 の場合、pH 5、2、3、20、28、および 21 ~ 3 に対してそれぞれ 3.5、4.2、5、8、3、および 8 枚の画像を使用しました。 DpHF19 の場合、pH 7、8、8、28、4、および 5 ~ 3 に対してそれぞれ 3.5、4.2、5、8、3、および 8 枚の画像を使用しました。 DpHF19_9his の場合、pH 6、6、8、14、15、8、および 4 ~ 3 の場合はそれぞれ 3.5、4.2、5、6、8、3、および 8 つの画像を使用しました。
クライオEM
クライオ EM サンプルは、Vitrobot (ThermoFisher) を使用して CFLAT ホーリーカーボン グリッドにタンパク質を塗布し、液体を吸い取り、グリッドを液体エタンに浸すことによって調製されました。 DpHF19 のビデオは、カウント モードで動作する K-2 Summit Direct Detect カメラ (Gatan Inc.) を備えた Glacios 顕微鏡 (ThermoFisher) で、ピクセルあたり 1.16 Å のピクセル サイズ、50 フレーム、総電子線量で取得されました。 65Åの - 2。 DpHF18 および DpHF7 のビデオは、ピクセルあたり 2 Å のピクセル サイズ、0.525 フレームの超解像度モードで動作する K-50 Summit Direct Detect カメラ (Gatan Inc.) を備えた Titan Krios (ThermoFisher) で取得されました。総電子線量 90 Å - 2。 Leginon を使用して自動データ収集を実行しました19 バージョン3.4。データ処理はcryoSPARCを使用して実行されました8、ワークフローは補足図にまとめられています。 10–12。ビデオはパッチ モーション補正によって調整され、超解像度ビデオは 1.05 Å のピクセル サイズにビニングされました。コントラスト伝達関数 (CTF) パラメーターは、パッチ CTF を使用して推定されました。テンプレートフリーのフィラメント トレースを画像のサブセットに対して実行し、結果として得られた粒子を 2D 分類にかけました。次に、選択した 2D クラスを、完全なデータセットに対するテンプレートベースのフィラメント トレースのテンプレートとして使用しました。複数回の 2D 分類の後、選択された粒子は、らせん対称性が課せられ、不均一な精製が可能になった 3D 精製を受けました。 DpHF19 では、7 スタート ヘリカル対称パラメーターではなく、個々の非接触サブユニットに関連する 19 スタート ヘリカル対称性を課しました。 DpHFXNUMX と DpHFXNUMX では、粒子ごとのデフォーカス、ビームチルト、球面収差も改善されました。密度変更は Phenix の ResolveCryoEM を使用して実行されました20,21 バージョン phenix-1.20.1。 DpHF18 および DpHF19 の原子モデルは ISOLDE を使用してクライオ EM マップに洗練されました22、その後、回転異性体とラマチャンドラン拘束を無効にし、入力開始モデルによって課せられた参照拘束を使用して、Phenix で実空間の洗練が行われます。 DpHF7 のモデルの解明には、セグメント化されたクライオ EM 非対称ユニット密度に関する de novo モデル構築プロトコルが使用されました。23。その後の残留物の取り込みと精製は、RosettaCM を使用して達成されました。24 バージョン 2019.31 では、セグメント化されていないクライオ EM マップ全体の対称性を利用して、密度とフィラメント内インターフェイスの最適な適合を実現します。 DpHF18 および DpHF19 について前述したように、実空間リファインメントの最終ラウンドは Phenix で実行されました。 Cryo-EM データの収集、改良、および検証の統計は補足表にまとめられています。 1.
TIRFM
ファイバーアセンブリ
pH 応答性線維の播種された核形成を画像化するために、DpHF18 線維を 488 つの異なるマレイミド結合蛍光団、Oregon5 および sulfo-Cy10 で標識しました。繊維を、PBS + 1 mM TCEP中で室温で4時間、25倍モル過剰で標識した後、ZebaスピンカラムでTBS (100 mM Tris、8.0 mM NaCl、pH 30)に緩衝液交換し、30 μMに濃縮した。 30μMの緑色繊維を、1Mクエン酸塩(繊維20μlに対してクエン酸塩0.6μl)を添加してpHを3.0に下げることによって分解した。溶液を5分間インキュベートした後、トリス(1Mストック3.6μl)を添加してpHを8.0に戻した。 1μMの組み立てられたDpHF0.6-Cy20ファイバー3.0μlを溶液に添加した。続いて溶液を室温でインキュベートした後、5 で遠心分離しました。 g 卓上遠心分離機で2分間。繊維をTBSに再懸濁し、TIRFMによって画像化しました。
ファイバーの分解
低 pH で分解する繊維の高速 TIRFM イメージングは、高速レンズと完璧なフォーカス システムを備えた Nikon Ti スタンドをベースにした特注の TIRF システムで実行されました。 Z ピエゾステージ (ASI)、カスタム拡張視野 (Cairn) および PLAN Apo 2 NA ×1.45 対物レンズを備えた方位角 TIRF 照明装置 (iLas100、Roper France)。画像は、方位角照明と同期した擬似グローバル シャッター モードで実行される Photometrics Prime 95B 裏面照射型 sCMOS カメラで取得されました。システムはMetamorph 7.10.1.161で動作しました。 Sulfo-Cy5 マレイミド標識ファイバーは、630 nm レーザー (Cairn レーザー発射に取り付けられた 150 mW Coherent OBIS) でイメージングされ、Cairn Optospin ホイールに取り付けられた Chroma ET655lp フィルターを使用して、1 ms ごとに 16 フレームのフレーム レートでイメージングされました。
線維は、クリーンルームグレードのカバースリップ (カスタム、25 × 8.0 mm) に取り付けられた Ibidi フローセル内のイメージングバッファー (100 mM Tris pH 25、75 mM NaCl) 中でイメージングされました。2、Nexterion)、PLL-PEG(0.1 mg ml )で不動態化-1 20 mM Hepes、pH 7.6 中で。 5分)。結合していない繊維をイメージングバッファーで除去する前に、繊維をカバースリップ上に 5 分間堆積させました。高速取得中に、低 pH 緩衝液 (25 mM Tris、100 mM NaCl、pH 3.0) を流すことによって pH を下げました。
バルク溶液中での繊維の分解を測定するために、1.5 ml エッペンドルフチューブ内の予め形成された繊維をより低い pH のクエン酸緩衝液に交換して分解を刺激しました。各 pH 反応の一部をさまざまな時点で取り出し、96 ウェル プレートに添加し、10 分間繊維を沈降させてガラス基板に付着させました。各条件および時点について、2500 つの視野を IN Cell Analyzer 60HS 顕微鏡 (Molecular Devices) で Nikon ×0.95 PLAN Apo 631 NA 空気対物レンズおよび 150 nm LED 励起源を使用し、684 ms の露光時間で収集した発光で取得しました。 24 ± XNUMX nm バンドパス フィルターを介して。カスタム CellProfiler スクリプトを使用して画像を定量化し、Otsu 閾値アルゴリズムで繊維をセグメント化しました。25。閾値の上限と下限、およびオブジェクト ID の適応ウィンドウは、ファイバーがバックグラウンド信号と比較して正しく識別されるまで調整されました。 CellProfiler パイプラインを使用して特定されたオブジェクトの長軸の長さを、各 pH 条件のインキュベーション時間に対してプロットしました。
液相AFM
試料調製
新しく劈開した白雲母雲母表面 (10 mm、Ted Pella Inc.) 上で 0.01 wt% ポリリジン溶液 12 μl を 2 分間インキュベートしました。過剰な溶液を除去し、表面を水ですすぎ、N で乾燥させました。2 ガス7。次に、イメージングバッファー(30 mM Tris-HCl、10 mM NaCl、pH 25)中の400μMタンパク質溶液8μlをポリリジンでコーティングされた雲母上で30分間インキュベートし、イメージバッファーで洗浄して過剰なタンパク質を除去しました。分解バッファー (25 mM Tris-HCl、400 mM NaCl、pH 4.1、4.4、4、5、または 4.7) の pH を 10 M NaOH または 1 M クエン酸で調整し、使用前に孔径 0.1 μm の PVDF フィルターで濾過しました。 。光酸実験では、10 mM Tris-HCl pH 25 中の 8 μM タンパク質溶液を裸の雲母上で 30 分間インキュベートし、25 mM Tris-HCl pH 5.5 で洗浄しました。表面上の繊維の数密度が低い場合は、追加の堆積およびリンスステップが実行されました。また、1 mM Tris-HCl pH 2 で 25 mM 5.5-ニトロベンズアルデヒド (Sigma-Aldrich) を新たに調製し、どの段階でも光にさらさずにすぐに使用しました。26。分光学的測定と pH 測定により、2-ニトロベンズアルデヒドは 200 ~ 405 nm の波長で活性化可能であり、pH を 5.5 から 2.7 に低下させること、およびレーザー強度が高いほど消費と酸性化が速くなることが示されました。
イメージング
一定の組成での速度論的研究のために、タンパク質でコーティングされたポリリジン雲母基板を AFM 液体セル (Bruker Multimode8) の下に置きました。画像は、きれいな窒化ケイ素カンチレバー (Bruker、SNL-10、バネ定数: 0.12 N m) を使用してイメージング バッファーにキャプチャされました。-1、室温 (5 °C) でタッピングモードで 25 分間 UV オゾン処理)。分解バッファーを流す前に、パラメーター (10 走査線、256 Hz 走査速度、高積分ゲイン (1.5 ~ 3)、および 4 ~ 50 mV 自由振幅) を最適化するために、ファイバーを 100 分間連続してイメージングしました。カンチレバーによる損傷が発生していないことを確認した後、分解バッファーを 25 µl min で連続注入しました。-1。フロースルーのセットアップは、滞留時間を無視し、素早い pH 切り替えを実現するように最適化されました。10.
光酸の研究では、25 mM Tris-HCl pH 5.5 を含むタンパク質でコーティングされた雲母を、通気バルブ付きの BlueDrive レーザー (×0.3 強度フィルター、波長 405 nm) を備えた Cypher VRS AFM (Asylum Research) の液体セルの下に置きました。開いてタッピングモードで操作します。繊維の表面被覆率が高いことを確認した後、イメージングバッファーを 1 mM Tris-HCl pH 2 中の 25 mM 5.5-ニトロベンズアルデヒドに置き換え、可視背景光にさらさずに操作し、再度イメージングしました。次にカンチレバーを後退させ、BlueDrive をオンにして、AFM の電動光学顕微鏡を使用して、事前に選択した領域全体を繰り返しラスター表示しました。スポットおよびライン パターンのラスター/ドウェル中の合計 UV 露光時間は 10 分を超えず、その後カンチレバーを露光領域に戻してイメージングしました。全体的な pH 変化については、光酸溶液と接触している AFM 液体セルの石英窓を手持ちの UV ランプ (波長 364 nm) に 7 分間曝露し、画像化しました。
画像は Gwyddion SPM v2.62 データ分析ソフトウェアで処理され、Fiji ソフトウェア v1.53s で分析されました。18。速度論については、繊維の全長が測定され、すでに分解されているとみなされる断片は長さの測定から除外されました。個々のファイバーの各端での分解率を測定するには(補足図) 8)、繊維の中心 (最初の長さの半分) が長さを測定するための 2 番目の端として割り当てられましたが、繊維断片の場合は、断片の中心が 2 番目の端として測定されました。
- SEO を活用したコンテンツと PR 配信。 今日増幅されます。
- PlatoData.Network 垂直生成 Ai。 自分自身に力を与えましょう。 こちらからアクセスしてください。
- プラトアイストリーム。 Web3 インテリジェンス。 知識増幅。 こちらからアクセスしてください。
- プラトンESG。 カーボン、 クリーンテック、 エネルギー、 環境、 太陽、 廃棄物管理。 こちらからアクセスしてください。
- プラトンヘルス。 バイオテクノロジーと臨床試験のインテリジェンス。 こちらからアクセスしてください。
- 情報源: https://www.nature.com/articles/s41565-024-01641-1

