Proteiner driver många biologiska processer, såsom enzymaktivitet, molekylär transport och cellulärt stöd. Den tredimensionella strukturen hos ett protein ger insikt i dess funktion och hur det interagerar med andra biomolekyler. Experimentella metoder för att bestämma proteinstruktur, såsom röntgenkristallografi och NMR-spektroskopi, är dyra och tidskrävande.
Däremot kan nyligen utvecklade beräkningsmetoder snabbt och exakt förutsäga strukturen av ett protein från dess aminosyrasekvens. Dessa metoder är kritiska för proteiner som är svåra att studera experimentellt, såsom membranproteiner, målen för många läkemedel. Ett välkänt exempel på detta är AlphaFold, en djupinlärningsbaserad algoritm hyllad för sina exakta förutsägelser.
ESMFold är en annan mycket exakt, djupinlärningsbaserad metod utvecklad för att förutsäga proteinstruktur från dess aminosyrasekvens. ESMFold använder en stor proteinspråksmodell (pLM) som ryggrad och fungerar från början till slut. Till skillnad från AlphaFold2 behöver den ingen uppslagning eller Multipelsekvensjustering (MSA) steg, och det förlitar sig inte heller på externa databaser för att generera förutsägelser. Istället tränade utvecklingsteamet modellen på miljontals proteinsekvenser från UniRef. Under träningen utvecklade modellen uppmärksamhetsmönster som elegant representerar de evolutionära interaktionerna mellan aminosyror i sekvensen. Denna användning av en pLM istället för en MSA möjliggör upp till 60 gånger snabbare förutsägelsetider än andra toppmoderna modeller.
I det här inlägget använder vi den förtränade ESMFold-modellen från Hugging Face with Amazon SageMaker att förutsäga den tunga kedjestrukturen av trastuzumab, en monoklonal antikropp utvecklades först av Genentech för behandling av HER2-positiv bröstcancer. Att snabbt förutsäga strukturen av detta protein kan vara användbart om forskare ville testa effekten av sekvensändringar. Detta kan potentiellt leda till förbättrad patientöverlevnad eller färre biverkningar.
Det här inlägget ger ett exempel på Jupyter-anteckningsboken och relaterade skript i det följande GitHub repository.
Förutsättningar
Vi rekommenderar att du kör det här exemplet i en Amazon SageMaker Studio anteckningsbok kör den PyTorch 1.13 Python 3.9 CPU-optimerade bilden på en ml.r5.xlarge-instanstyp.
Visualisera trastuzumabs experimentella struktur
Till att börja använder vi biopython bibliotek och ett hjälpskript för att ladda ner trastuzumab-strukturen från RCSB Protein Data Bank:
Därefter använder vi py3Dmol bibliotek för att visualisera strukturen som en interaktiv 3D-visualisering:
Följande figur representerar 3D-proteinstrukturen 1N8Z från Protein Data Bank (PDB). I den här bilden visas trastuzumabs lätta kedja i orange, den tunga kedjan är blå (med den variabla regionen i ljusblått) och HER2-antigenet är grönt.

Vi kommer först att använda ESMFold för att förutsäga strukturen av den tunga kedjan (kedja B) från dess aminosyrasekvens. Sedan kommer vi att jämföra förutsägelsen med den experimentellt bestämda strukturen som visas ovan.
Förutsäg trastuzumabs tunga kedjestruktur från dess sekvens med hjälp av ESMFold
Låt oss använda ESMFold-modellen för att förutsäga strukturen hos den tunga kedjan och jämföra den med experimentresultatet. Till att börja med använder vi en förbyggd bärbar datormiljö i Studio som kommer med flera viktiga bibliotek, som PyTorch, förinstallerad. Även om vi skulle kunna använda en accelererad instanstyp för att förbättra prestandan för vår anteckningsbokanalys, använder vi istället en icke-accelererad instans och kör ESMFold-förutsägelsen på en CPU.
Först laddar vi den förtränade ESMFold-modellen och tokenizern från Kramar Face Hub:
Därefter kopierar vi modellen till vår enhet (CPU i det här fallet) och ställer in några modellparametrar:
För att förbereda proteinsekvensen för analys måste vi tokenisera den. Detta översätter aminosyrasymbolerna (EVQLV...) till ett numeriskt format som ESMFold-modellen kan förstå (6,19,5,10,19,...):
Därefter kopierar vi den tokeniserade ingången till läget, gör en förutsägelse och sparar resultatet i en fil:
Detta tar cirka 3 minuter på en icke-accelererad instanstyp, som en r5.
Vi kan kontrollera noggrannheten i ESMFold-förutsägelsen genom att jämföra den med den experimentella strukturen. Vi gör detta med hjälp av US-Align verktyg utvecklat av Zhang Lab vid University of Michigan:
| PDB-kedja1 | PDB-kedja2 | TM-poäng |
| data/prediction.pdb:A | data/experimentell.pdb:B | 0.802 |
Smakämnen mall modellering poäng (TM-poäng) är ett mått för att bedöma likheten mellan proteinstrukturer. En poäng på 1.0 indikerar en perfekt matchning. Poäng över 0.7 indikerar att proteiner delar samma ryggradsstruktur. Poäng över 0.9 indikerar att proteinerna är funktionellt utbytbara för nedströms användning. I vårt fall att uppnå TM-Score 0.802, skulle ESMFold-förutsägelsen sannolikt vara lämplig för tillämpningar som strukturpoäng eller ligandbindningsexperiment, men kanske inte lämplig för användningsfall som molekylär ersättning som kräver extremt hög noggrannhet.
Vi kan validera detta resultat genom att visualisera de inriktade strukturerna. De två strukturerna visar en hög, men inte perfekt, grad av överlappning. Proteinstrukturförutsägelser är ett område i snabb utveckling och många forskarlag utvecklar allt mer exakta algoritmer!
Distribuera ESMFold som en SageMaker slutpunkt
Att köra modellinferens i en bärbar dator är bra för experiment, men vad händer om du behöver integrera din modell med en applikation? Eller en MLOps pipeline? I det här fallet är ett bättre alternativ att distribuera din modell som en slutpunkt för slutledning. I följande exempel kommer vi att distribuera ESMFold som en SageMaker slutpunkt i realtid på en accelererad instans. SageMaker realtidsslutpunkter ger ett skalbart, kostnadseffektivt och säkert sätt att distribuera och vara värd för maskininlärningsmodeller (ML). Med automatisk skalning kan du justera antalet instanser som kör slutpunkten för att möta kraven i din applikation, optimera kostnaderna och säkerställa hög tillgänglighet.
Den förbyggda SageMaker behållare för Hugging Face gör det enkelt att distribuera modeller för djupinlärning för vanliga uppgifter. Men för nya användningsfall som förutsägelse av proteinstruktur måste vi definiera en anpassad inference.py skript för att ladda modellen, köra förutsägelsen och formatera utdata. Det här skriptet innehåller mycket av samma kod som vi använde i vår anteckningsbok. Vi skapar också en requirements.txt fil för att definiera några Python-beroenden för vår slutpunkt att använda. Du kan se filerna vi skapade i GitHub repository.
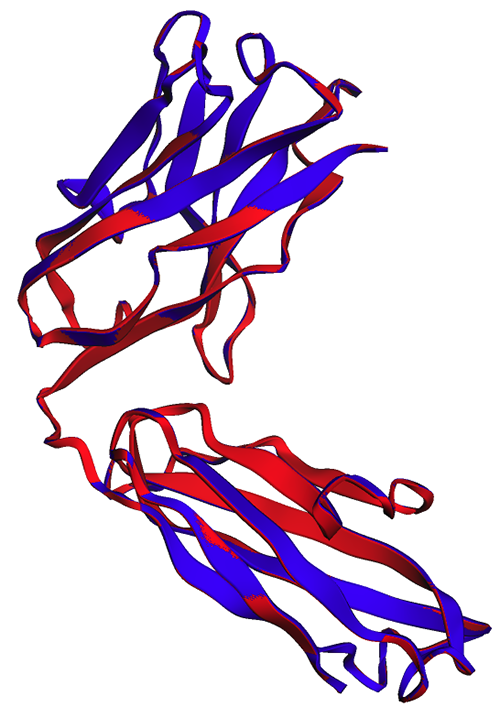
I följande figur är de experimentella (blå) och förutsagda (röda) strukturerna för den tunga trastuzumabkedjan mycket lika, men inte identiska.

Efter att vi har skapat de nödvändiga filerna i code katalog distribuerar vi vår modell med SageMaker HuggingFaceModel klass. Detta använder en förbyggd behållare för att förenkla processen att distribuera Hugging Face-modeller till SageMaker. Observera att det kan ta 10 minuter eller mer att skapa slutpunkten, beroende på tillgängligheten av ml.g4dn instanstyper i vår region.
När slutpunktsdistributionen är klar kan vi skicka in proteinsekvensen igen och visa de första raderna i förutsägelsen:
Eftersom vi distribuerade vår slutpunkt till en accelererad instans bör förutsägelsen bara ta några sekunder. Varje rad i resultatet motsvarar en enda atom och inkluderar aminosyraidentiteten, tre rumsliga koordinater och en pLDDT poäng representerar förutsägelseförtroendet på den platsen.
| PDB_GROUP | ID | ATOM_LABEL | RES_ID | CHAIN_ID | SEQ_ID | CARTN_X | CARTN_Y | CARTN_Z | BETALNING | PLDDT | ATOM_ID |
| ATOM | 1 | N | G.L.U. | A | 1 | 14.578 | -19.953 | 1.47 | 1 | 0.83 | N |
| ATOM | 2 | CA | G.L.U. | A | 1 | 13.166 | -19.595 | 1.577 | 1 | 0.84 | C |
| ATOM | 3 | CA | G.L.U. | A | 1 | 12.737 | -18.693 | 0.423 | 1 | 0.86 | C |
| ATOM | 4 | CB | G.L.U. | A | 1 | 12.886 | -18.906 | 2.915 | 1 | 0.8 | C |
| ATOM | 5 | O | G.L.U. | A | 1 | 13.417 | -17.715 | 0.106 | 1 | 0.83 | O |
| ATOM | 6 | cg | G.L.U. | A | 1 | 11.407 | -18.694 | 3.2 | 1 | 0.71 | C |
| ATOM | 7 | cd | G.L.U. | A | 1 | 11.141 | -18.042 | 4.548 | 1 | 0.68 | C |
| ATOM | 8 | OE1 | G.L.U. | A | 1 | 12.108 | -17.805 | 5.307 | 1 | 0.68 | O |
| ATOM | 9 | OE2 | G.L.U. | A | 1 | 9.958 | -17.767 | 4.847 | 1 | 0.61 | O |
| ATOM | 10 | N | VAL | A | 2 | 11.678 | -19.063 | -0.258 | 1 | 0.87 | N |
| ATOM | 11 | CA | VAL | A | 2 | 11.207 | -18.309 | -1.415 | 1 | 0.87 | C |
Med samma metod som tidigare ser vi att anteckningsboken och slutpunktsförutsägelserna är identiska.
| PDB-kedja1 | PDB-kedja2 | TM-poäng |
| data/endpoint_prediction.pdb:A | data/prediction.pdb:A | 1.0 |
Som observerats i följande figur visar ESMFold-förutsägelserna som genereras i den bärbara datorn (röd) och av slutpunkten (blå) perfekt inriktning.
Städa upp
För att undvika ytterligare avgifter tar vi bort våra slutpunkts- och testdata:
Sammanfattning
Förutsägelse av beräkningsproteinstruktur är ett viktigt verktyg för att förstå proteiners funktion. Utöver grundforskning har algoritmer som AlphaFold och ESMFold många tillämpningar inom medicin och bioteknik. De strukturella insikter som genereras av dessa modeller hjälper oss att bättre förstå hur biomolekyler interagerar. Detta kan sedan leda till bättre diagnostiska verktyg och terapier för patienter.
I det här inlägget visar vi hur man distribuerar ESMFold-proteinspråksmodellen från Hugging Face Hub som en skalbar slutpunkt med SageMaker. För mer information om hur du distribuerar Hugging Face-modeller på SageMaker, se Använd Hugging Face med Amazon SageMaker. Du kan också hitta fler proteinvetenskapliga exempel i Fantastisk proteinanalys på AWS GitHub repo. Lämna gärna en kommentar om det finns några andra exempel du vill se!
Om författarna
 Brian Loyal är senior AI/ML Solutions Architect i Global Healthcare and Life Sciences-teamet på Amazon Web Services. Han har mer än 17 års erfarenhet av bioteknik och maskininlärning, och brinner för att hjälpa kunder att lösa genomiska och proteomiska utmaningar. På fritiden tycker han om att laga mat och äta med sina vänner och familj.
Brian Loyal är senior AI/ML Solutions Architect i Global Healthcare and Life Sciences-teamet på Amazon Web Services. Han har mer än 17 års erfarenhet av bioteknik och maskininlärning, och brinner för att hjälpa kunder att lösa genomiska och proteomiska utmaningar. På fritiden tycker han om att laga mat och äta med sina vänner och familj.
 Shamika Ariyawansa är en AI/ML Specialist Solutions Architect i Global Healthcare and Life Sciences-teamet på Amazon Web Services. Han arbetar passionerat med kunder för att påskynda deras AI och ML adoption genom att tillhandahålla teknisk vägledning och hjälpa dem att förnya och bygga säkra molnlösningar på AWS. Utanför jobbet älskar han skidåkning och terrängåkning.
Shamika Ariyawansa är en AI/ML Specialist Solutions Architect i Global Healthcare and Life Sciences-teamet på Amazon Web Services. Han arbetar passionerat med kunder för att påskynda deras AI och ML adoption genom att tillhandahålla teknisk vägledning och hjälpa dem att förnya och bygga säkra molnlösningar på AWS. Utanför jobbet älskar han skidåkning och terrängåkning.
 Yanjun Qi är Senior Applied Science Manager på AWS Machine Learning Solution Lab. Hon förnyar och tillämpar maskininlärning för att hjälpa AWS-kunder att påskynda deras AI- och molnintroduktion.
Yanjun Qi är Senior Applied Science Manager på AWS Machine Learning Solution Lab. Hon förnyar och tillämpar maskininlärning för att hjälpa AWS-kunder att påskynda deras AI- och molnintroduktion.
- SEO-drivet innehåll och PR-distribution. Bli förstärkt idag.
- PlatoAiStream. Web3 Data Intelligence. Kunskap förstärkt. Tillgång här.
- Minting the Future med Adryenn Ashley. Tillgång här.
- Köp och sälj aktier i PRE-IPO-företag med PREIPO®. Tillgång här.
- Källa: https://aws.amazon.com/blogs/machine-learning/accelerate-protein-structure-prediction-with-the-esmfold-language-model-on-amazon-sagemaker/

