The NMPA granted innovation approvals to Medtronic’s renal denervation device and issued a review report.
This is the sixth report published this year for imported devices, after those from Medtronic (Implantable deep brain stimulation directional lead), CarboFix Orthopedics, Intuitive Surgical, Abbott and Ivantis.
The published review reports like this one serve as important references for you to understand what the regulatory authorities are thinking and evaluating during their review process. We have been following the list for the past several years and review the relevant ones for our clients’ specific products to gain more clarity and be more efficient in their submission and approval process. As NMPA standardizes and streamlines the review process for fast-track approval, domestic and overseas players can benefit from our expertise and experience.
Product overview
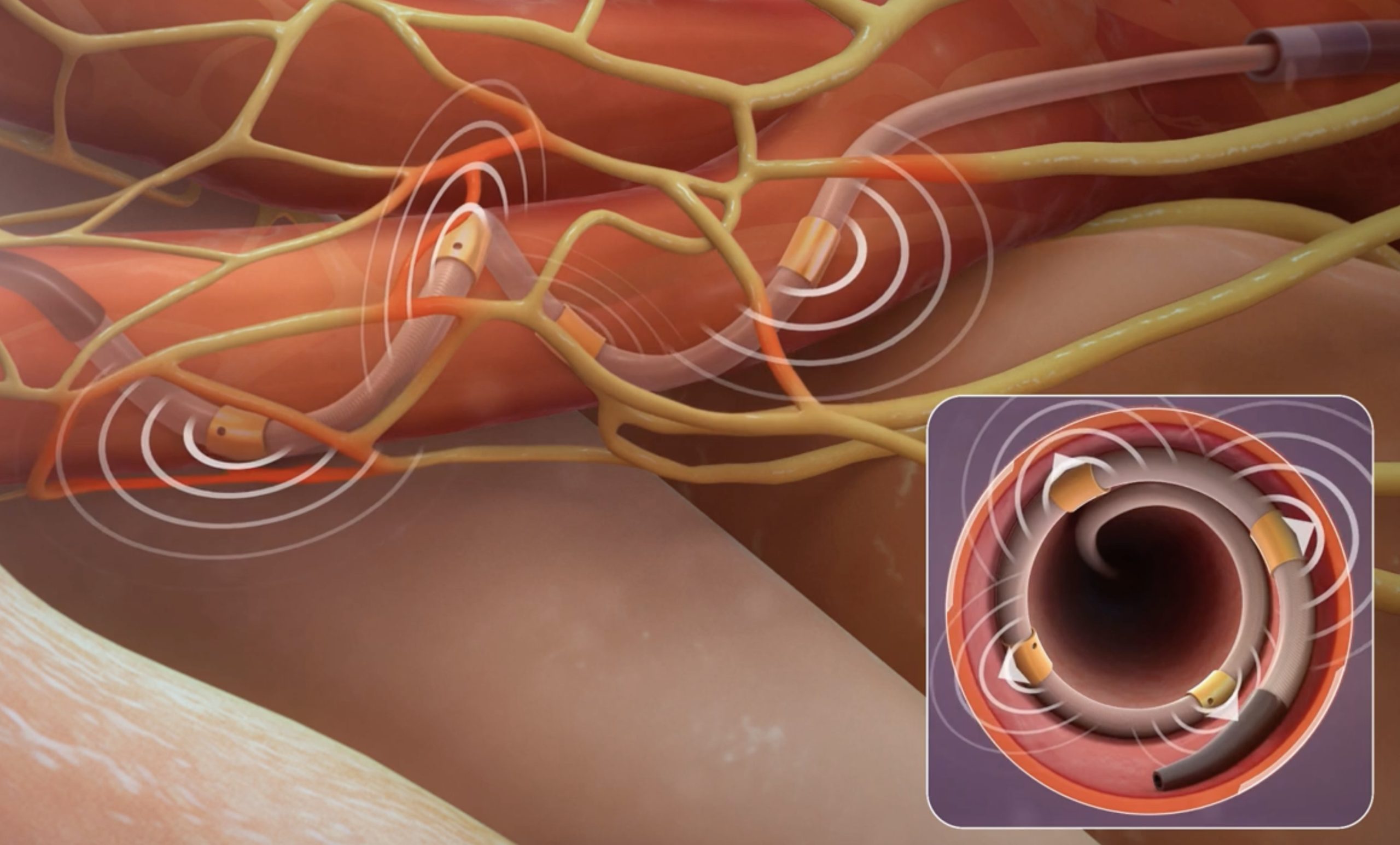
(I)Product structure and composition
(II)Intended Use
The product is intended for use in medical institutions and is designed to be used in conjunction with the renal artery radiofrequency ablation device produced by Medtronic (Model: RDNG3A, Software Release Version: 2). It is suitable for assisting in the treatment of resistant hypertension and hypertension in patients who are intolerant to medication.
Resistant hypertension is defined as patients whose blood pressure is not adequately controlled despite the use of three or more antihypertensive drugs (including a diuretic) for more than three months.
Medication intolerance refers to patients who cannot tolerate medication due to contraindications or adverse drug reactions.
(III)Model/Specification
(IV)Working principle
Pre-clinical
(I) Product Performance Research
The applicant has provided documentation on product performance research and the development of product technical requirements, outlining the basis for determining functional and safety indicators such as physical properties, chemical properties, sterility, bacterial endotoxins, electrical performance, temperature measurement accuracy, electrical safety, and electromagnetic compatibility. The technical requirements for the product reference relevant national and industry standards, including YY 0285.1-2017 and YY 0778-2018.
(II) Biocompatibility
The applicant evaluated the biocompatibility of the catheter, which directly contacts the patient, according to GB/T 16886.1-2011. The evaluation materials have brief contact with the human circulatory system and included biological tests (cytotoxicity, sensitization, intradermal reaction, acute systemic toxicity, pyrogen, hemolysis, complement activation, coagulation, in vivo thrombosis). A biological test report issued by an overseas testing institution was submitted, meeting the relevant requirements.
(III) Sterilization
The product is sterilized by the manufacturing enterprise through irradiation, with a sterility assurance level of 10^-6. The applicant has specified the sterilization method and parameters, and sterilization verification was conducted according to ISO 11137-1 standards. A sterilization verification report was submitted, and residual toxicity studies are not involved.
(IV) Product Shelf Life and Packaging
The product is for single use, with a shelf life of 3 years. The applicant conducted accelerated aging tests to verify the shelf life, testing the product and packaging under equivalent aging conditions. Post-aging tests on temperature measurement accuracy, power accuracy, working length, and sterility showed results that meet the requirements. The applicant also provided research data related to storage and transportation.
(V) Animal Studies
The applicant provided a total of four animal study reports:
- PS629 Study: A 28-day non-GLP study using a pig model aimed to verify the difference in use between the submitted product and the previous generation, and to confirm the product’s safety and theoretical efficacy. Compared to the control group, comprehensive parameters were used to determine the device’s safety and potential efficacy. Results indicated all test animals survived to the end point, and the device was successfully delivered to the target renal artery without major surgery or device-related complications. Treated animals showed significantly lower norepinephrine levels than the control group, suggesting preliminary efficacy.
- PS716 and PS717 Studies: 7-day and 28-day non-GLP studies using a pig model to evaluate a treatment strategy combining treatment of renal artery branches and the main renal artery. Four different treatment methods were used, targeting various positions in the renal artery for single or combined treatment. Safety was judged by histopathological examination, and efficacy was evaluated by examining the morphology and physiological effects on renal nerves. Results showed no clinically significant complications in treated vessels, adjacent structures, or kidneys. By days 7 and 28, the combined treatment significantly reduced renal cortical axon density and norepinephrine levels compared to the control group, demonstrating efficacy.
- FS235 Study: A 180-day GLP study using a pig model to assess the long-term safety and efficacy of the product. Results showed successful procedures with no major surgery or device-related complications. At 180 days, treated vessels showed no significant new intimal formation, surgical damage, dissection, aneurysm formation, or significant arterial dilation. Histologically, treated vessels showed complete re-endothelialization at 180 days with no adverse changes in the vessel wall or surrounding organs. The norepinephrine concentration and terminal axon density in the cortical renal tissue were significantly lower compared to the control group, indicating sustained treatment effects over 180 days.
(VI) Safety Indicators of Active Devices
The product complies with the general requirements of GB 9706.1-2020, the specific safety requirements of GB 9706.202-2021, and the parallel safety requirements for electromagnetic compatibility of YY 9706.102-2021. Inspection reports from medical device testing institutions, conducted in conjunction with our company’s renal artery radiofrequency ablation device, were provided.
(VII) Others
The applicant submitted analysis and research data on factors influencing the treatment efficacy of the product. Based on relevant animal and clinical trial data, factors such as ablation point positioning, number of ablation points, operator experience, and learning curve were analyzed. It was concluded that under the standardized operating procedures, these factors do not significantly impact the final treatment efficacy.
The applicant provided technical documentation on RFID, including interoperability research with the host software, and submitted usability study data for the product.
Clinical
The clinical evaluation of the product was conducted through clinical trials, with the applicant submitting three sets of overseas clinical data, including two clinical trials and one clinical experience dataset. The details are as follows:
_Clinical design
1. OFF MED Trial
The OFF MED trial employed a Bayesian adaptive design in a prospective multicenter randomized controlled study. It included exploratory and confirmatory phases, with sample sizes adjusted based on interim analyses to ensure appropriate power.
Participants: 80 in the exploratory phase and 251 in the confirmatory phase, with a 1:1 randomization ratio, resulting in a combined cohort of 331 participants.
Intervention: Sham surgery as control, with patients off antihypertensive medication from 3-4 weeks before the procedure until 3 months post-treatment.
Sample Size Calculation: Simulated 15,000 times to ensure posterior probability ≥ 0.975, controlling Type I error within a one-sided 0.025 threshold.
Endpoints:
Primary Safety: Major Adverse Events (MAE) within one month post-randomization.
Primary Efficacy: Change in ambulatory systolic blood pressure (ASBP) at 3 months.
Secondary Efficacy: Changes in clinic systolic blood pressure (SBP), diastolic blood pressure (DBP), and other time points up to 24 months.
2. ON MED Trial
The ON MED trial also used a Bayesian adaptive design in a prospective multicenter randomized controlled study.
Participants: 80 in the exploratory phase and 257 in the confirmatory phase, with randomization ratios of 1:1 and 2:1 respectively.
Intervention: Sham surgery as control, targeting hypertensive patients with poorly controlled blood pressure on medication.
Sample Size Calculation: Similar to OFF MED, with interim analyses guiding the sample size.
Endpoints:
Primary Safety: MAE within one month post-randomization.
Primary Efficacy: Change in ASBP at 6 months.
Secondary Efficacy: Changes in SBP, DBP, and ABPM readings at various intervals up to 24 months.
_Trial Results
Safety Evaluation: Combined safety analysis from OFF MED and ON MED trials was conducted on the first 253 enrolled subjects.
Safety Endpoint: MAE rate within one-month post-procedure. The target performance goal (PG) was set at 7.1%, with the actual MAE incidence rate observed at 0.4%, significantly below the PG, confirming the safety of the procedure.
Efficacy Evaluation
The product demonstrated a significant reduction in ASBP and SBP in hypertensive patients:
OFF MED Trial: Average ASBP reduction at 3 months.
ON MED Trial: Sustained ASBP reduction at 6 months, with additional secondary endpoints confirming long-term efficacy.
_Conclusion
The OFF MED and ON MED clinical trials, combined with GSR study data, met the primary safety performance endpoints and demonstrated the product’s efficacy in reducing ASBP and SBP in hypertensive patients, affirming its clinical benefit and safety.
Please email us at info@ChinaMedDevice.com to see if NMPA released review reports for your device. We can translate for you with nominal fees.
- SEO Powered Content & PR Distribution. Get Amplified Today.
- PlatoData.Network Vertical Generative Ai. Empower Yourself. Access Here.
- PlatoAiStream. Web3 Intelligence. Knowledge Amplified. Access Here.
- PlatoESG. Carbon, CleanTech, Energy, Environment, Solar, Waste Management. Access Here.
- PlatoHealth. Biotech and Clinical Trials Intelligence. Access Here.
- Source: https://chinameddevice.com/medtronic-renal-denervation-device/

