Het duurt ongeveer 10 tot 15 jaar om een medicijn van de bank naar het bed te krijgen, en ongeveer 6 tot 7 van die jaren worden besteed aan klinische onderzoeken. Hoewel het moeilijk is om het exacte bedrag te bepalen, lijken de kosten voor de ontwikkeling van geneesmiddelen te variëren van minder dan $1 miljard tot ruim $2 miljard AMERIKAANSE DOLLAR. Hier volgen de stappen die een medicijn neemt op weg naar goedkeuring, met een diepgaande evaluatie van de vier klinische onderzoeksfasen en hoe deze passen in het goedkeuringsproces voor geneesmiddelen.
Doelstellingen van klinisch onderzoek
Klinische onderzoeken kijken naar nieuwe manieren om ziekten te voorkomen, op te sporen of te behandelen.
Klinische onderzoeken kunnen het volgende onderzoeken:
- Nieuwe medicijnen of nieuwe combinaties van medicijnen
- Nieuwe manieren om een operatie uit te voeren
- Nieuwe medische apparaten
- Nieuwe manieren om bestaande behandelingen te gebruiken
- Nieuwe manieren om gedrag te veranderen om de gezondheid te verbeteren
- Nieuwe manieren om de levenskwaliteit van mensen met acute of chronische ziekten te verbeteren
Gemeenschappelijke doelen voor onderzoeken met een experimenteel medicijn:
- Ontdek of het het gewenste effect heeft (doet echt wat we denken dat het doet)
- Bepaal de meest effectieve en veilige dosering
- Ontdek bijwerkingen en zorg ervoor dat het risico op de bijwerkingen niet groter is dan de te behandelen ziekte
- Ontdek hoe een medicijn in het lichaam wordt afgebroken en hoe lang het in het lichaam blijft
- Bepaal welke voedingsmiddelen, dranken, kruiden of andere medicijnen tegelijkertijd met het experimentele medicijn kunnen worden gebruikt en wat moet worden vermeden omdat dit een bijwerking zou kunnen veroorzaken
Clinical Trials
Dankzij de resultaten van klinische onderzoeken kunnen de FDA en regelgevende instanties in andere landen beslissen of een medicijn of apparaat veilig en effectief is bij mensen en goedgekeurd moet worden om op de markt te komen.
De medicijnen en apparaten die in fase 0-3 van klinische onderzoeken worden gebruikt, zijn niet goedgekeurd voor het beoogde gebruik, maar de klinische onderzoeken worden uitgevoerd onder toezicht van de regerende regelgevende instantie (FDA in de VS) of op basis van hun criteria.
Een medicijn kan als ‘nieuw’ worden beschouwd, zelfs als het al jaren in gebruik is, op voorwaarde dat er een verandering wordt voorgesteld in het gebruik, de verpakking, de formulering, de toedieningsweg of het gebruik in de patiëntenpopulatie waarbij het risico zou toenemen.
Jaren geleden heeft de FDA bijvoorbeeld een medicijn goedgekeurd om hoge bloeddruk te behandelen. De fabrikant van het medicijn wil het nu testen als behandeling tegen angst bij volwassenen. Dit nieuwe gebruik van het medicijn zou als experimenteel worden beschouwd.
Pad naar goedkeuring van medicijnen
Wanneer een sponsor (geneesmiddelontwikkelaar) een nieuw middel ontwikkelt en goedkeuring wil aanvragen, zijn er verschillende stappen die het nieuwe medicijn moet doorlopen om aan te tonen dat het veilig en effectief is.

Preklinische onderzoeken zijn de eerste stap. Bij dit soort onderzoek wordt gebruik gemaakt van celculturen of dieren om de voorlopige werkzaamheid, toxiciteit, farmacokinetiek (hoe het medicijn in het lichaam werkt) en veiligheidsinformatie te bepalen. Als de resultaten veelbelovend zijn, kunnen klinische onderzoeken beginnen.
Onderzoek Nieuw Drug (IND)
Een sponsor die een klinische proef wil uitvoeren met een nieuw medicijn (zoals een medicijn, vaccin of biologisch product waarvoor goedkeuring wordt gevraagd door de FDA) moet een Investigational New Drug-aanvraag (IND) indienen bij de FDA. De FDA beoordeelt de IND-aanvraag voor de veiligheid van het gebruik van de medicijnen bij mensen en om te verzekeren dat de voorgestelde klinische onderzoeken geen onredelijk risico voor menselijke proefpersonen opleveren. De FDA verifieert ook dat er sprake is van adequate geïnformeerde toestemming en bescherming van menselijke proefpersonen.
Het volgende moet door de FDA worden ingediend en beoordeeld voordat een IND wordt toegewezen:
- Resultaten van dierproeven tonen aan dat het medicijn redelijk veilig is voor gebruik bij mensen
- Klinische protocollen (studieplannen) voor het uitvoeren van klinische onderzoeken
- Ervaring met het medicijn in andere landen en gegevens uit eerder onderzoek op mensen
- Productie-informatie
- Afhankelijkheid en misbruikpotentieel
- Informatie over de onderzoeker die toezicht houdt op het onderzoek
In de VS, klinische proeven kan pas starten nadat de IND is beoordeeld door de FDA en een protocol is beoordeeld en goedgekeurd door een Institutional Review Board (IRB).
Een IRB is een onafhankelijk orgaan dat is opgericht om de rechten en het welzijn van deelnemers aan menselijk onderzoek te beschermen. IRB's zorgen ervoor dat het onderzoek aanvaardbaar is, dat deelnemers toestemming hebben gegeven en volledig op de hoogte zijn van hun risico's, en dat onderzoekers passende stappen ondernemen om patiënten tegen schade te beschermen.
Buiten de VS worden onderzoeken op dezelfde manier beoordeeld door een IRB of ethische commissie die specifiek is voor hun land of regio.
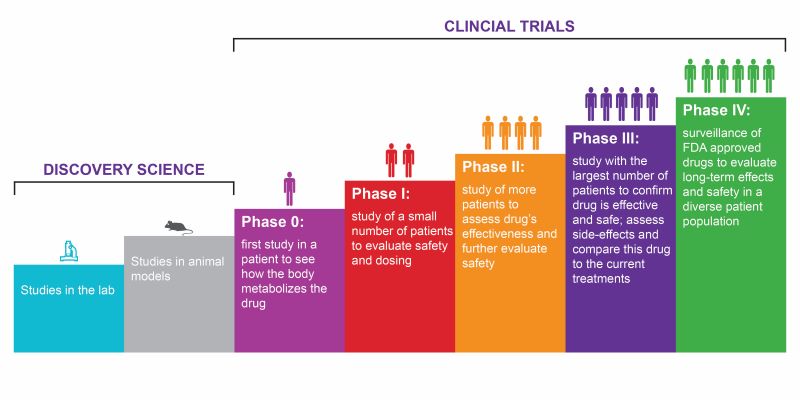
Het pad naar goedkeuring: klinische onderzoeksfasen
Nieuwe behandelingen doorlopen de volgende fasen van klinisch onderzoek:
Fase O-onderzoeken
Eerste klinische proef bij mensen (optioneel) met 10 tot 15 gezonde vrijwilligers, waarbij kleine hoeveelheden van het onderzoeksgeneesmiddel worden toegediend om te controleren of het medicijn zich gedraagt zoals verwacht bij mensen. Als de medicatie anders werkt dan verwacht, zal er hoogstwaarschijnlijk aanvullend preklinisch onderzoek worden afgerond voordat wordt besloten of er verder wordt gegaan.
Fase I-onderzoeken
Onderzoekers testen een medicijn of behandeling bij 20 tot 80 mensen (vaak een first-in-human onderzoek), vaak gezonde vrijwilligers, om te zien of het medicijn of de behandeling werkt. veilig, bijwerkingen identificeren en veilige doseringsbereiken bepalen. Bij het werven van patiënten voor deze fase gaat het vaak om het vinden van gezonde vrijwilligers die bereid zijn strenge tests te ondergaan tijdens hun verblijf in de onderzoeksfaciliteit.
Fase II-onderzoeken
Het onderzoeksgeneesmiddel wordt aan een grotere groep (honderden) mensen gegeven die lijden aan de betreffende ziekte. Dit is om het te bepalen effectiviteit en om de veiligheid, bijwerkingen op korte termijn en doseringen verder te bestuderen. Soms zijn fase 2-studies verdeeld in fase 2a en 2b:
- 2a (proof of concept) – bepaal het werkingsmechanisme van het medicijn en hoe het medicijn het lichaam beïnvloedt om de werkzaamheid aan te tonen bij mensen met de doelziekte. Als de werkzaamheid niet veelbelovend blijkt te zijn, zullen er geen verdere tests plaatsvinden.
- 2b (dosis-responsonderzoeken) – bepaal de optimale dosering(en) voor verdere onderzoeken.
Deelnemers aan fase II-onderzoek moeten de doelziekte hebben, maar niet veel comorbiditeiten. De in- en uitsluitingscriteria zijn restrictiever, waardoor rekrutering een uitdaging wordt. Er is een gericht wervingsplan nodig.
Fase III-onderzoeken
Het onderzoeksgeneesmiddel of de experimentele behandeling wordt aan grote groepen mensen (duizenden) gegeven bevestig de effectiviteit ervan, controleer bijwerkingen, vergelijk het met standaard of soortgelijke behandelingenen informatie verzamelen waardoor het nieuwe medicijn of de nieuwe behandeling veilig kan worden gebruikt. Soms worden fase 3-onderzoeken uitgevoerd als 3a en 3b:
- 3a (cruciale onderzoeken) – bevestigen de veiligheid en effectiviteit van een grotere populatie en ondersteunen initiële claims van de sponsor bij regelgevende instanties. Deze fase wordt uitgevoerd voordat de eerste indicatiegoedkeuring wordt verkregen (er is aangetoond dat het medicijn veilig en effectief is voor het beoogde gebruik).
- 3b (na voltooiing van 3a) – verzamel aanvullende gegevens, zoals het beste gebruik van het medicijn in de praktijk, subpopulaties van patiënten, langetermijnresultaten of effecten op de kwaliteit van leven. Deze fase begint na het verkrijgen van de eerste goedgekeurde indicatie van de regelgevende instantie en vóór het verkrijgen van goedkeuring voor het in de handel brengen met nauwkeurige etikettering van het geneesmiddel.
Fase III-studie Inclusie- en exclusiecriteria zijn minder restrictief, maar er is een groot aantal patiënten nodig, vaak uit verschillende landen. Er is een krachtig mondiaal wervingsplan nodig.
Fase IV-onderzoeken
Nadat een medicijn door de FDA is goedgekeurd en voor het publiek beschikbaar is gesteld, volgen onderzoekers het medicijn veiligheid en effectiviteit bij de algemene bevolking (duizenden deelnemers), op zoek naar meer informatie over de voordelen van een medicijn, het optimale gebruik en de langetermijneffecten.
Nieuwe medicijnaanvraag (NDA)
De FDA heeft een beoordelingsvergadering met de medicijnsponsor voordat een geheimhoudingsverklaring wordt ingediend. De NDA is het vehikel waarmee medicijnsponsors formeel voorstellen dat de FDA een nieuw geneesmiddel voor verkoop en marketing in de VS goedkeurt. De gegevens verzameld tijdens preklinische en fase 0-3 van klinische onderzoeken van een IND worden onderdeel van de NDA. Om goedkeuring te verkrijgen, moet de NDA voldoende informatie verstrekken zodat de FDA-reviewers kunnen bepalen of:
- Het medicijn is veilig en effectief voor het voorgestelde gebruik
- De voordelen van het medicijn wegen zwaarder dan de risico's
- De voorgestelde etikettering van het geneesmiddel (bijsluiter) is passend
- De methoden die worden gebruikt bij de vervaardiging van het medicijn en de controles die worden gebruikt om de kwaliteit van het medicijn te behouden, zijn adequaat om de identiteit, kracht, kwaliteit en zuiverheid van het medicijn te behouden
De documentatie die vereist is in een geheimhoudingsverklaring wordt verondersteld het hele verhaal van het medicijn te vertellen. Dit omvat wat er gebeurde tijdens de klinische onderzoeksfasen van het testen, de ingrediënten van het medicijn, de resultaten van de dierstudies, hoe het medicijn zich in het lichaam gedraagt en hoe het wordt vervaardigd, verwerkt en verpakt.
Zodra de NDA is ingediend, heeft de FDA 60 dagen de tijd om te bepalen of deze compleet genoeg is voor beoordeling. Als de NDA ter beoordeling wordt ingediend, beoordeelt een FDA-team de veiligheid en werkzaamheid van het onderzoek dat door de sponsor van het onderzoek is ingediend. Reviewers bepalen of zij het eens zijn met de resultaten en conclusies van de sponsor, of dat zij aanvullende informatie nodig hebben om te kunnen beslissen. FDA-inspecteurs reizen ook naar klinische onderzoekslocaties om routine-inspecties uit te voeren. Ze zoeken naar bewijs van verzinsel, manipulatie of het achterhouden van gegevens. Het beoordelingsteam brengt een aanbeveling uit en een hoge FDA-functionaris beslist.
Etikettering van medicijnen
Zodra de FDA besluit dat een medicijn veilig en effectief is voor het beoogde gebruik, werken ze samen met de sponsor aan een nauwkeurige etikettering van medicijnen. Etikettering omvat:
- Voorschrijfinformatie voor beroepsbeoefenaren in de gezondheidszorg
- Etikettering van dozen en containers
- Informatie voor patiënten of zorgverleners (bijv. Medicatiehandleidingen en bijsluiters voor patiënten)
De FDA inspecteert de fabrieken waar het medicijn wordt vervaardigd. Zij zullen de NDA goedkeuren of een antwoordbrief sturen.
Er zijn snellere manieren om goedkeuring te verkrijgen voor geneesmiddelen die ernstige of levensbedreigende ziekten behandelen of in een onvervulde behoefte voorzien. Meer informatie is beschikbaar op https://www.fda.gov/drugs/development-approval-process-drugs. Bijvoorbeeld medicijnen die behandelen zeldzame ziekten of levensbedreigende diagnoses.
Fase IV klinische onderzoeken
Nadat een medicijn door de FDA is goedgekeurd en voor het publiek beschikbaar is gesteld, volgen onderzoekers de veiligheid en effectiviteit ervan bij de algemene bevolking. Dit gebeurt via fase IV klinische onderzoeken. De bedoeling van deze proeven is om:
- Zoek meer informatie over de voordelen van het medicijn
- Optimaal gebruik
- Zeldzamere bijwerkingen
- Lange-termijn effecten
Informatie verzameld uit fase IV-onderzoeken kan een verandering in de etikettering veroorzaken. Zoals het bijwerken van bijwerkingen, of gebruik in combinatie met andere ziekten/medicijnen. Ook kan verwijdering uit de markt plaatsvinden als er te veel risico wordt gedetecteerd.
Het imperiale voordeel in alle klinische onderzoeksfasen
Elke klinische onderzoeksfase vereist unieke strategieën voor het werven en inschrijven van patiënten. Imperial Clinical Research Services heeft ruime ervaring met het ontwikkelen, vertalen en afdrukken van wervingsmateriaal voor alle klinische onderzoeksfasen voor gebruik in mondiale onderzoeken. Bovendien houdt Imperial je studie op koers vermijd vertragingen.
Dan McDonald's blog over studie opstarten en uitvoeren bespreekt waarom “het van cruciaal belang is om die eerste patiënt op tijd in het onderzoek te krijgen. Als dat doel niet wordt bereikt, duurt het langer om grip te krijgen en is de kans groter dat u uw inschrijvingsdoelen voor klinische onderzoeken niet zult halen. Je onderzoek zal niet aan de gestelde termijnen voldoen.”
We helpen ook met lokale vereisten. De meeste fase 3-onderzoeken zijn internationaal. Verschillen in lokale vereisten van land tot land maken multinationale studies bijzonder uitdagend. Om het onderzoek op lokaal niveau uit te voeren is maatwerk nodig. Dit omvat onderhoud, kalibratie, verwijdering, douane-inklaring en importregelgeving, technologische infrastructuur, gereguleerde apparaten, gebruiksinstructies en vertalingen.
Erica Manning herinnert ons eraan in haar vertaalblog voor klinische proeven dat “Nauwkeurige vertaaldiensten voor klinische onderzoeken de sleutel zijn tot het algehele succes van een onderzoek. Elke deelnemer moet dezelfde informatie in zijn moedertaal ontvangen om consistente onderzoeksberichten in alle talen en nauwkeurigheid van het protocol te garanderen.”
Het kwaliteitsmanagementsysteem van Imperial Translation Services is door Intertek gecertificeerd volgens ISO 9001:2015. Dit toont aan dat u zich inzet voor de kwaliteit van uw vertalingen van klinische onderzoeken.
Algemene inlichtingen voor meer informatie over hoe we tijd en geld kunnen besparen voor uw klinische onderzoeken.
- Door SEO aangedreven content en PR-distributie. Word vandaag nog versterkt.
- PlatoData.Network Verticale generatieve AI. Versterk jezelf. Toegang hier.
- PlatoAiStream. Web3-intelligentie. Kennis versterkt. Toegang hier.
- PlatoESG. Automotive / EV's, carbon, CleanTech, Energie, Milieu, Zonne, Afvalbeheer. Toegang hier.
- BlockOffsets. Eigendom voor milieucompensatie moderniseren. Toegang hier.
- Bron: https://www.imperialcrs.com/blog/2023/08/08/drug-approval-process-four-clinical-research-phases/

