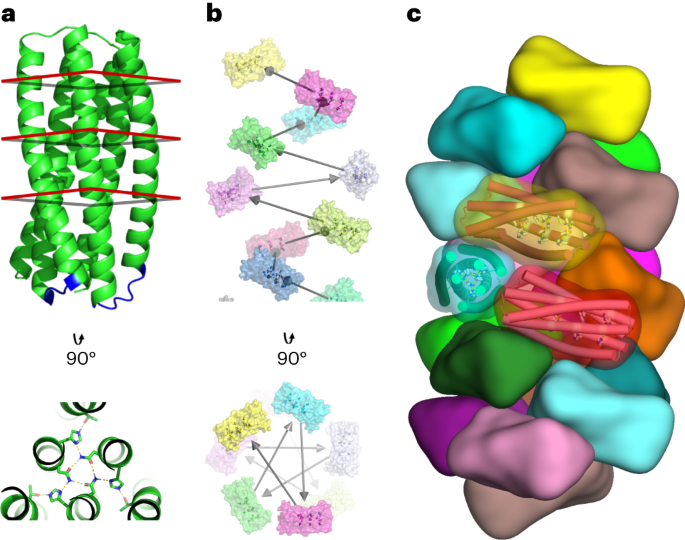
Computergestützte Designstrategie
Kurze Schleifen zur Verbindung von pRO-2.3-Helices zu einer einzigen Kette wurden unter Verwendung einer umfassenden Datenbank von Rückgratproben entworfen, die aus Fragmenten bestehen, die sich über zwei helikale Regionen erstrecken, wie durch DSSP in hochauflösenden kristallographischen Strukturen identifiziert (wie zuvor beschrieben).14). Schleifen wurden in dieser Datenbank durch starre Ausrichtung der terminalen Reste des Fragments und des Ziels mithilfe eines optimierten Überlagerungsalgorithmus identifiziert15. Kandidaten, die eine Ausrichtungstoleranz von 0.35 Å RMSD einhielten, wurden über Torsionsraumkoordinaten und weiche Koordinatenbeschränkungen für die ausgerichteten Schweratomkoordinaten des Kandidatenrückgrats auf das Zielrückgrat ausgerichtet. Kandidaten-Loop-Sequenzen wurden dann unter Sequenzprofilbeschränkungen entworfen, die durch Ausrichtung des Loop-Backbones an der Quellstrukturdatenbank generiert wurden. Die Kandidaten mit den niedrigsten Punktzahlen wurden für den Entwurf der Endschleife ausgewählt.
Helix-Andock- und Entwurfsmethoden7 wurden auf den verknüpften pRO-2.3 angewendet, um Designmodelle für helikale Filamente zu generieren. Die folgenden Kriterien filterten einzelne Entwurfsverläufe: eine Diskrepanz von mehr als −15.0 Rosetta-Energieeinheiten zwischen den gebundenen (polymeren) und ungebundenen (monomeren) Zuständen, eine Grenzflächenoberfläche von mehr als 700 Å2, eine Komplementarität der Rosetta-Form von mehr als 0.62 und eine Anzahl nicht erfüllter polarer Reste unter 5. Designs, die diese Kriterien erfüllten, wurden einer manuellen Verfeinerung unterzogen, wobei einzelne Umkehrungen zu Mutationen vorgenommen wurden, die nicht zur Stabilisierung des gebundenen Zustands der Schnittstelle beitragen. Das am besten bewertete Design für jede angedockte Konfiguration wurde dann zur experimentellen Validierung in einen endgültigen Proteinsatz integriert.
Proteinexpression und -reinigung
Die synthetischen Gene für insgesamt 18 Designs wurden für die Expression in optimiert Escherichia coli und von IDT erworben, dann in die multiple Klonierungsstelle des pET29b+-Vektors zwischen NdeI- und XhoI-Restriktionsstellen eingefügt. Diese Konstrukte wurden in BL21* (DE3) eingeführt. E. coli kompetente Zellen. Transformanten wurden in 50 ml Terrific Broth-Medium, ergänzt mit 200 mg l, kultiviert-1 Kanamycin. Die Expression erfolgte unter der Kontrolle eines T7-Promotors 24 Stunden lang bei 37 °C unter Verwendung der Studier-Autoinduktion16 bis die Kulturen durch Zentrifugation geerntet wurden. Zellpellets wurden in Tris-gepufferter Kochsalzlösung (TBS) resuspendiert und mit Bugbuster-Detergens lysiert. Die durch Zentrifugation geklärte lösliche Fraktion wurde einer Reinigung über Ni unterzogen2+ immobilisierte Metallaffinitätschromatographie unter Verwendung von Ni-NTA Superflow-Harz. Das Harz mit gebundenem Zelllysat wurde mit zehn Säulenvolumina 40 mM Imidazol und 500 mM NaCl gewaschen, gefolgt von der Elution mit 400 mM Imidazol und 75 mM NaCl. Die löslichen und unlöslichen Fraktionen wurden einer SDS-Polyacrylamid-Gelelektrophoreseanalyse unterzogen. Für das elektronenmikroskopische Screening wurden Proben ausgewählt, die Proteinbanden mit dem richtigen Molekulargewicht aufwiesen. Ausgewählte Designs wurden zur weiteren Charakterisierung auf 0.5 l skaliert, wobei die Expression erneut 24 Stunden lang bei 37 °C unter Verwendung der Studier-Autoinduktion erfolgte16 vor der Ernte durch Zentrifugation. Zellpellets wurden in TBS resuspendiert und durch Mikrofluidisierung lysiert, gefolgt von der oben beschriebenen Reinigung.
Negativer EM-Fleck
Lösliche Fraktionen wurden für das elektronenmikroskopische Screening in TBS (25 mM Tris-Puffer, 75 mM NaCl, pH 8) konzentriert. Ein 6-µl-Tropfen (1 µl Probe sofort mit 5 µl Puffer verdünnt) wurde auf negativ glimmentladene, kohlenstoffbeschichtete 200-Mesh-Kupfergitter aufgetragen, mit Milli-Q-Wasser gewaschen und entweder mit 0.75 % Uranylformiat (pH 4.0) gefärbt ) oder Nano-W (pH 6.8), erworben von Nanoprobes, Inc., wie zuvor beschrieben17. Das Screening wurde entweder mit einem 100-kV-Transmissionselektronenmikroskop (FEI) Morgagni M268 oder einem 120-kV-Transmissionselektronenmikroskop Talos L120C (ThermoFisher) durchgeführt. Die Bilder wurden mit einem unten montierten Teitz CMOS 4k-Kamerasystem aufgenommen und mit der Fiji-Software (Version: 2.14.0/1.54f) für einen verbesserten Kontrast verarbeitet.18 zur Klarheit.
Die Faserlängen wurden mithilfe des Faserverfolgungsalgorithmus in cryoSPARC quantifiziert8. Diese Methode identifiziert Fasern durch Kreuzkorrelation mit einer Template-Klasse und die Verfolgung zusammenhängender Fasern aus den identifizierten Partikeln. Für alle gemessenen Fasern wurde eine aus DpHF19 generierte Template-Klasse verwendet. Fasern wurden entsprechend der durchschnittlichen Krümmung (<0.0005 Å) gefiltert-1) und die durchschnittliche normalisierte Kreuzkorrelation (>0.5) über jede Faser. Für DpHF18 verwendeten wir 5, 2, 3, 20, 28 und 21 Bilder für pH 3, 3.5, 4.2, 5, 8 bzw. 3 bis 8. Für DpHF19 verwendeten wir 7, 8, 8, 28, 4 und 5 Bilder für pH 3, 3.5, 4.2, 5, 8 bzw. 3 bis 8. Für DpHF19_9his verwendeten wir 6, 6, 8, 14, 15, 8 und 4 Bilder wurden für pH 3, 3.5, 4.2, 5, 6, 8 bzw. 3 bis 8 verwendet.
Kryo-EM
Kryo-EM-Proben wurden hergestellt, indem Protein auf CFLAT-Lochkohlenstoffgitter aufgetragen, Flüssigkeit abgetupft und die Gitter mit einem Vitrobot (ThermoFisher) in flüssiges Ethan getaucht wurden. Für DpHF19 wurden Videos mit einem Glacios-Mikroskop (ThermoFisher) aufgenommen, das mit einer K-2 Summit Direct Detect-Kamera (Gatan Inc.) ausgestattet war und im Zählmodus arbeitete, mit einer Pixelgröße von 1.16 Å pro Pixel, 50 Bildern und einer Gesamtelektronendosis von 65 Å-2. Für DpHF18 und DpHF7 wurden Videos auf einem Titan Krios (ThermoFisher) aufgenommen, der mit einer K-2 Summit Direct Detect-Kamera (Gatan Inc.) ausgestattet war, die im Superauflösungsmodus mit einer Pixelgröße von 0.525 Å pro Pixel, 50 Bildern und arbeitet eine Gesamtelektronendosis von 90 Å-2. Die automatisierte Datenerfassung wurde mit Leginon durchgeführt19 Version 3.4. Die Datenverarbeitung wurde mit cryoSPARC durchgeführt8und Arbeitsabläufe sind in den ergänzenden Abbildungen zusammengefasst. 10-12. Die Videos wurden durch Patch-Bewegungskorrektur ausgerichtet, wobei hochauflösende Videos auf eine Pixelgröße von 1.05 Å gruppiert wurden. Die Parameter der Kontrastübertragungsfunktion (CTF) wurden mithilfe der Patch-CTF geschätzt. An einer Teilmenge der Bilder wurde eine schablonenfreie Filamentverfolgung durchgeführt und die resultierenden Partikel einer 2D-Klassifizierung unterzogen. Ausgewählte 2D-Klassen wurden dann als Vorlagen für die vorlagenbasierte Filamentverfolgung in vollständigen Datensätzen verwendet. Nach mehreren Runden der 2D-Klassifizierung wurden ausgewählte Partikel einer 3D-Verfeinerung unterzogen, wobei eine helikale Symmetrie eingeführt und eine ungleichmäßige Verfeinerung ermöglicht wurde. Für DpHF19 haben wir eine eingängige Helixsymmetrie eingeführt, die einzelne, nicht kontaktierende Untereinheiten in Beziehung setzt, und nicht die Parameter der zweigängigen Helixsymmetrie. Für DpHF7 und DpHF19 wurden auch die Defokussierung pro Partikel, die Strahlneigung und die sphärische Aberration verfeinert. Die Dichteänderung wurde mit ResolveCryoEM in Phenix durchgeführt20,21 Version phenix-1.20.1. Atommodelle für DpHF18 und DpHF19 wurden mithilfe von ISOLDE zu Kryo-EM-Karten verfeinert22, gefolgt von einer Realraumverfeinerung in Phenix, mit deaktivierten Rotamer- und Ramachandran-Beschränkungen und mit Referenzbeschränkungen, die durch das Eingabe-Startmodell auferlegt werden. Die Aufklärung des Modells für DpHF7 verwendete das De-novo-Modellbildungsprotokoll für die segmentierte asymmetrische Kryo-EM-Einheitsdichte23. Die anschließende Einarbeitung und Verfeinerung der Rückstände erfolgte mit RosettaCM24 Version 2019.31, die die Symmetrie der unsegmentierten Kryo-EM-Karte für optimale Anpassung an die Dichte und Intrafilament-Schnittstellen nutzt. Eine letzte Runde der Realraumverfeinerung wurde in Phenix durchgeführt, wie oben für DpHF18 und DpHF19 beschrieben. Statistiken zur Kryo-EM-Datenerfassung, -verfeinerung und -validierung sind in der Ergänzungstabelle zusammengefasst 1.
TIRFM
Fasermontage
Um die Keimbildung von pH-responsiven Fasern abzubilden, wurden DpHF18-Fasern mit zwei verschiedenen Maleimid-konjugierten Fluorophoren, Oregon488 und Sulfo-Cy5, markiert. Die Fasern wurden mit einem 10-fachen molaren Überschuss in PBS + 1 mM TCEP für 4 Stunden bei Raumtemperatur markiert, bevor der Puffer in TBS (25 mM Tris, 100 mM NaCl, pH 8.0) auf einer Zeba-Spin-Säule ausgetauscht und auf 30 μM konzentriert wurde . Grüne Fasern mit 30 μM wurden durch Zugabe von 1 M Citrat (0.6 μl Citrat auf 20 μl Fasern) zerlegt, um den pH-Wert auf 3.0 zu senken. Die Lösung wurde 5 Minuten lang inkubiert, bevor Tris (3.6 μl 1 M Stammlösung) zugegeben wurde, um den pH-Wert wieder auf 8.0 zu bringen; Der Lösung wurde 1 μl zusammengesetzte DpHF18-Cy5-Fasern mit 30 μM zugesetzt. Die Lösung wurde anschließend bei Raumtemperatur inkubiert und dann bei 13,000 zentrifugiert g für 2 Minuten in einer Tischzentrifuge. Die Fasern wurden in TBS resuspendiert und mit TIRFM abgebildet.
Demontage der Faser
Die schnelle TIRFM-Bildgebung von Fasern, die sich bei niedrigem pH-Wert zerlegen, wurde mit einem speziell angefertigten TIRF-System durchgeführt, das auf einem Nikon Ti-Stativ basiert und mit einem perfekten Fokussystem sowie einem schnellen ausgestattet ist Z Piezotisch (ASI), ein azimutaler TIRF-Illuminator (iLas2, Roper France) mit einem individuell erweiterten Sichtfeld (Cairn) und ein PLAN Apo 1.45 NA ×100-Objektiv. Die Bilder wurden mit einer von hinten beleuchteten sCMOS-Kamera Photometrics Prime 95B im Pseudo-Global-Shutter-Modus aufgenommen, synchronisiert mit der azimutalen Beleuchtung. Das System wurde von Metamorph 7.10.1.161 betrieben. Mit Sulfo-Cy5-Maleimid markierte Fasern wurden mit einem 630-nm-Laser (150 mW Coherent OBIS, montiert in einem Cairn-Laserstart) und mit einem Chroma ET655lp-Filter, montiert in einem Cairn Optospin-Rad, mit einer Bildrate von 1 Bild alle 16 ms abgebildet.
Die Fasern wurden in Bildgebungspuffer (25 mM Tris pH 8.0, 100 mM NaCl) in einer Ibidi-Durchflusszelle abgebildet, die auf Deckgläsern in Reinraumqualität (kundenspezifisch, 25 × 75 mm) montiert war2, Nexterion) und mit PLL-PEG (0.1 mg ml) passiviert-1 in 20 mM Hepes, pH 7.6; 5 Minuten). Die Fasern konnten sich 5 Minuten lang auf dem Deckglas ablagern, bevor ungebundene Fasern mit dem Bildpuffer entfernt wurden. Während der schnellen Erfassung wurde der pH-Wert durch Einleiten von Puffer mit niedrigem pH-Wert (25 mM Tris, 100 mM NaCl, pH 3.0) gesenkt.
Um die Faserzerlegung in Massenlösung zu messen, wurden vorgeformte Fasern in 1.5-ml-Eppendorf-Röhrchen in Citratpuffer bei niedrigerem pH-Wert ausgetauscht, um die Zerlegung zu stimulieren. Ein Teil jeder pH-Reaktion wurde zu verschiedenen Zeitpunkten entnommen und 96 Minuten lang in eine Platte mit 10 Vertiefungen gegeben, damit sich die Fasern absetzen und am Glassubstrat haften konnten. Für jede Bedingung und jeden Zeitpunkt wurden neun Sichtfelder mit einem IN Cell Analyzer 2500HS-Mikroskop (Molecular Devices) unter Verwendung eines Nikon ×60 PLAN Apo 0.95 NA-Luftobjektivs und einer 631-nm-LED-Anregungsquelle bei einer Belichtungszeit von 150 ms mit erfasster Emission erfasst durch einen 684 ± 24 nm Bandpassfilter. Die Bilder wurden mithilfe eines benutzerdefinierten CellProfiler-Skripts quantifiziert, um Fasern mit dem Otsu-Schwellenwertalgorithmus zu segmentieren25. Ober- und Untergrenzen des Schwellenwerts sowie das adaptive Fenster für die Objekt-ID wurden angepasst, bis Fasern im Verhältnis zum Hintergrundsignal korrekt identifiziert wurden. Die Hauptachsenlänge der mithilfe der CellProfiler-Pipeline identifizierten Objekte wurde gegen die Inkubationszeit für jede pH-Bedingung aufgetragen.
Flüssigphasen-AFM
Probenvorbereitung
Wir inkubierten 10 µl einer 0.01 Gew.-%igen Polylysinlösung auf einer frisch gespaltenen Muskovit-Glimmeroberfläche (12 mm, Ted Pella Inc.) für 2 Minuten. Die überschüssige Lösung wurde entfernt und die Oberfläche mit Wasser gespült und mit N getrocknet2 Gas7. Dann wurden 30 µl 10 µM Proteinlösung im Bildpuffer (25 mM Tris-HCl, 400 mM NaCl bei pH 8) 30 Minuten lang auf dem mit Polylysin beschichteten Glimmer inkubiert und mit dem Bildpuffer gewaschen, um überschüssiges Protein zu entfernen. Der pH-Wert des Zerlegungspuffers (25 mM Tris-HCl, 400 mM NaCl, pH 4.1, 4.4, 4, 5 oder 4.7) wurde mit 10 M NaOH oder 1 M Zitronensäure eingestellt und vor der Verwendung mit einem PVDF-Filter mit 0.1 µm Porengröße filtriert . Für Photosäureexperimente wurde eine 10 µM Proteinlösung in 25 mM Tris-HCl, pH 8, 30 Minuten lang auf blankem Glimmer inkubiert und mit 25 mM Tris-HCl, pH 5.5, gewaschen; Bei geringer Faserdichte auf der Oberfläche wurde ein zusätzlicher Abscheidungs- und Spülschritt durchgeführt. Wir haben auch 1 mM 2-Nitrobenzaldehyd (Sigma-Aldrich) in 25 mM Tris-HCl, pH 5.5, frisch hergestellt und es sofort verwendet, ohne dass es zu irgendeinem Zeitpunkt Licht ausgesetzt wurde26. Spektroskopische und pH-Messungen zeigten, dass 2-Nitrobenzaldehyd zwischen Wellenlängen von 200 und 405 nm aktivierbar ist und den pH-Wert von 5.5 auf 2.7 senkt, und dass eine höhere Laserintensität zu einem schnelleren Verbrauch und einer schnelleren Versauerung führt.
Imaging
Für die kinetische Studie bei konstanter Zusammensetzung wurden die proteinbeschichteten Polylysin-Glimmersubstrate unter die AFM-Flüssigkeitszelle (Bruker Multimode8) gelegt. Die Bilder wurden im Bildpuffer mit einem sauberen Siliziumnitrid-Cantilever (Bruker, SNL-10, Federkonstante: 0.12 N·m) aufgenommen-1, 5 min UV-ozonisiert) im Zapfbetrieb bei Raumtemperatur (25 °C). Vor dem Fließen des Zerlegungspuffers wurden die Fasern 10 Minuten lang kontinuierlich abgebildet, um die Parameter zu optimieren (256 Scanlinien, 1.5 Hz Scanrate, hohe Integralverstärkung (3–4) und 50–100 mV freie Amplitude). Nachdem bestätigt wurde, dass keine durch den Cantilever verursachten Schäden auftraten, wurde der Zerlegungspuffer kontinuierlich mit 25 µl pro Minute injiziert-1. Der Durchflussaufbau wurde optimiert, um eine vernachlässigbare Verweilzeit und eine schnelle pH-Umschaltung zu ermöglichen10.
Für die Photosäurestudie wurde proteinbeschichteter Glimmer mit 25 mM Tris-HCl pH 5.5 unter die Flüssigkeitszelle eines Cypher VRS AFM (Asylum Research) gelegt, das mit einem BlueDrive-Laser (×0.3 Intensitätsfilter, 405 nm Wellenlänge) mit Entlüftungsventil ausgestattet war geöffnet und im Zapfbetrieb betrieben. Nachdem die hohe Oberflächenbedeckung der Fasern bestätigt wurde, wurde der Abbildungspuffer durch 1 mM 2-Nitrobenzaldehyd in 25 mM Tris-HCl, pH 5.5, ersetzt, ohne Einwirkung von sichtbarem Hintergrundlicht betrieben und erneut abgebildet. Anschließend wurde der Ausleger zurückgezogen, BlueDrive eingeschaltet und mithilfe des motorisierten optischen Mikroskops des AFM wiederholt über vorgewählte Bereiche gerastert. Die gesamte UV-Belichtungszeit während des Rasterns/Verweilens für Punkt- und Linienmuster betrug nicht mehr als 10 Minuten, danach wurde der Ausleger zurück zu den belichteten Bereichen bewegt und abgebildet. Für globale pH-Änderungen wurde das Quarzfenster der AFM-Flüssigkeitszelle in Kontakt mit der Photosäurelösung 364 Minuten lang einer tragbaren UV-Lampe (Wellenlänge 7 nm) ausgesetzt und dann abgebildet.
Die Bilder wurden mit der Datenanalysesoftware Gwyddion SPM v2.62 verarbeitet und mit der Fiji-Software v1.53s analysiert18. Für die Kinetik wurde die gesamte Faserlänge gemessen und alle Fragmente, die als bereits zerlegt galten, wurden von der Längenmessung ausgeschlossen. Um die Zerlegungsrate an jedem Ende einzelner Fasern zu messen (ergänzende Abb. 8) wurde die Mitte der Faser (die Hälfte der ursprünglichen Länge) als zweites Ende zur Längenmessung zugewiesen, während bei Faserfragmenten die Mitte des Fragments als zweites Ende gemessen wurde.
- SEO-gestützte Content- und PR-Distribution. Holen Sie sich noch heute Verstärkung.
- PlatoData.Network Vertikale generative KI. Motiviere dich selbst. Hier zugreifen.
- PlatoAiStream. Web3-Intelligenz. Wissen verstärkt. Hier zugreifen.
- PlatoESG. Kohlenstoff, CleanTech, Energie, Umwelt, Solar, Abfallwirtschaft. Hier zugreifen.
- PlatoHealth. Informationen zu Biotechnologie und klinischen Studien. Hier zugreifen.
- Quelle: https://www.nature.com/articles/s41565-024-01641-1

