Kwaliteitscontrole, afstemming en SNP-oproepen
De op ddRAD-sequencing gebaseerde genotypering van 58 individuen behorend tot zes inheemse runderrassen; Gir-, Sahiwal-, Tharparkar-, Rathi-, Red Sindhi- en Kankrej-runderen met hun geografische en ecologische verspreiding (fig. 1) inclusief het productieve doel, vachtkleur, representatieve agroklimatologische zone, broedgebied, de geografische coördinaat van elk broedgebied samen met dier-ID en geslacht van elk individu weergegeven in aanvullende tabel S1; resulteerde in 138.59 miljoen onbewerkte aflezingen, wat overeenkomt met 23 miljoen aflezingen per ras en 2.2 miljoen aflezingen per dier. Na initiële filtering op basis van leeskwaliteit en adapterverwijdering, bleef het merendeel van de leesbewerkingen (138.58 miljoen leesbewerkingen; 99.9%) behouden (aanvullende tabel S2). Een hoog percentage leesbewerkingen (94.53%) werd toegewezen aan de Bos taurus (ARS-UCD1.2) referentie-assemblage (aanvullende tabel S2). In deze studie is geprobeerd om alleen de SNP's van verschillende rundveerassen te analyseren, daarom werden alle andere varianten niet meegenomen in de daaropvolgende analyse. Het aantal SNP's in 6 runderrassen varieerde tussen 8,42,768 en 3,81,966 na individuele variantoproep. Het maximale aantal SNP's werd waargenomen in SAC (8,42,768), gevolgd door GIC (8,34,780), KAC (8,10,279), RAC (8,05,020), RSC (6,72,632) en THC (3,81,966) (Tafel 1). De gecombineerde dataset over 6 runderrassen leverde in totaal 43,47,445 SNP's op. Vervolgens werd het VCF-bestand stapsgewijs verwerkt om SNP's van lage kwaliteit eruit te filteren. Ten eerste werden de SNP's gefilterd op leesdiepte van 2 (RD 2), leesdiepte van 5 (RD 5) en leesdiepte van 10 (RD 10). Voor verdere analyse werd de dataset van 9,82,174 SNP's geïdentificeerd bij RD van 5 gebruikt voor daaropvolgende analyse (tabel 1). Al die SNP's die aanwezig waren met een lage dekking (RD <5) werden uit de dataset verwijderd. De SNP's die werden geïdentificeerd bij RD van 5 werden verder gefilterd met behulp van verschillende criteria, zoals het aandeel ontbrekende genotypen, de kleine allelfrequentie en Hardy Weinberg Equilibrium (HWE). De filterreeks resulteerde in een totaal van 84,027 SNP's van hoge kwaliteit. Na het filteren varieerde het aantal SNP's tussen de rassen aanzienlijk. Het hoogste aantal SNP's werd waargenomen in GIC (34,743), gevolgd door RSC (13,092), KAC (12,812), SAC (8956), THC (7356) en RAC (7068) (Tabel 2).
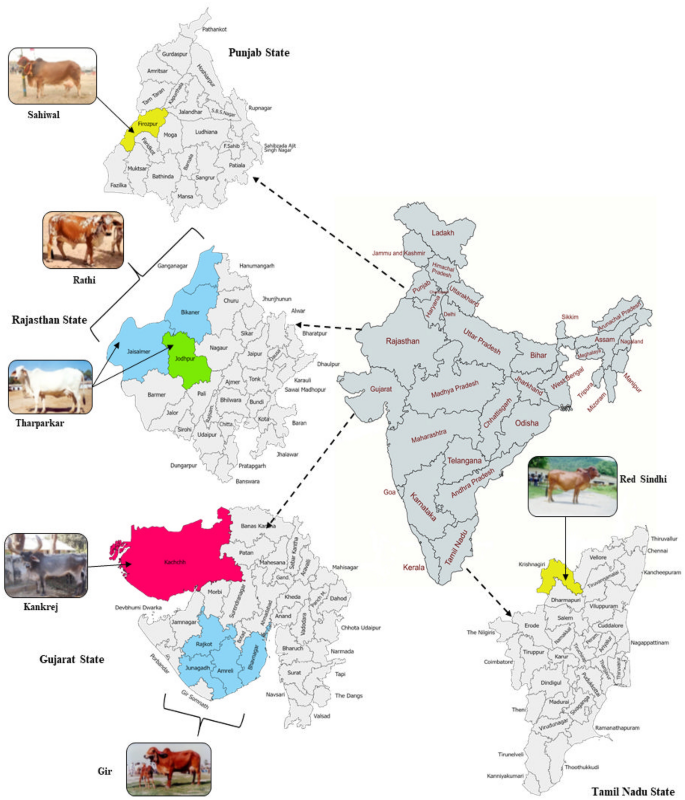
Geografische spreiding van zes runderrassen die in deze studie zijn opgenomen (de kaart is gegenereerd met behulp van websites Map Chart https://www.mapchart.net/ en verfkaarten https://paintmaps.com/).
Functionele annotatie van varianten
De samengevoegde hoogwaardige SNP-dataset van alle 6 melkrassen werd geannoteerd Bos taurus (ARS-UCD1.2) referentie genoom. Met betrekking tot hun verdeling in het genoom, werd voorspeld dat een groot aantal geannoteerde SNP's zich in het intronische gebied zouden bevinden (41,372 SNP's, 53.87%), gevolgd door intergene regio's (26,834 SNP's, 34.94%). Er waren slechts 948 SNP's (1.23%) die werden verspreid in de exonische regio's. Verder waren er 3497 SNP's (4.55%) gelokaliseerd binnen het gebied van 5 Kb stroomopwaarts en 3661 SNP's (4.77%) stroomafwaarts van de transcriptiestartplaats. De analyse resulteerde ook in 93 SNP's (0.121%) in 5'UTR, 293 SNP's (0.38%) in 3'UTR-regio. Er werd ook voorspeld dat in totaal 8 SNP's (0.01%) een voortijdig stopcodon zouden veroorzaken (Fig. 2).
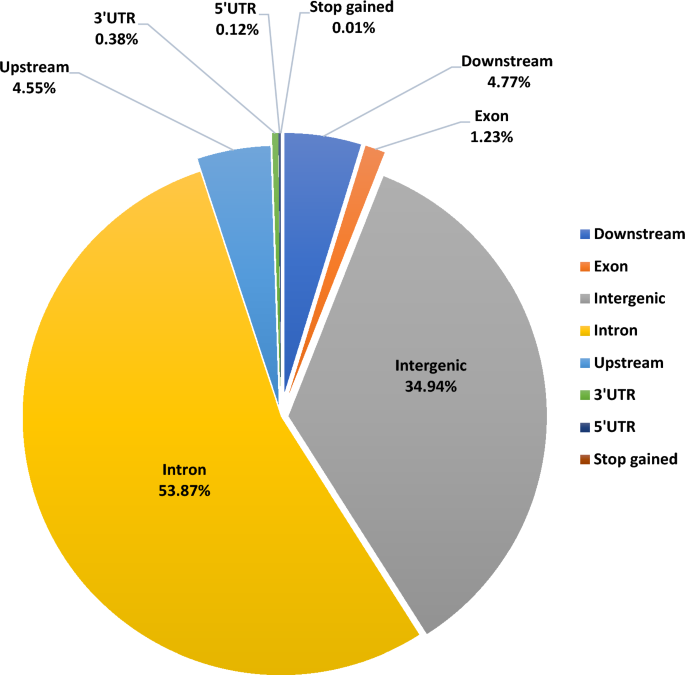
Algehele verdeling van SNP's met betrekking tot genomische distributie voor alle rassen.
Op basis van de impact van SNP's op eiwitcoderende genen, werden de SNP's gecategoriseerd als hebbende een hoge impact (10 SNP's; 0.01%), een matige impact (298 SNP's; 0.39%) en een lage impact (697 SNP's; 0.91%). . De meerderheid van de SNP's (75,801; 98.69%) werd geïdentificeerd als modifier (aanvullende tabel S3). Bovendien was een groot deel van de SNP's (65.74%) stil van aard, gevolgd door missense (33.37%) en onzin (0.89%), met een gemiddelde missense/silent-ratio van 0.507 (aanvullende tabel S4). Bovendien bleken van alle gesubstitueerde genotypen die in de huidige studie zijn geïdentificeerd, C / T- en G / A-genotypes de overhand te hebben, terwijl A / T-genotype in de laagste proporties bleek te zijn (aanvullende tabel S5). Voor individuele rassen zijn de annotatieresultaten samengevat in Fig. 3 en aanvullende tabel S6. In GIC werd voorspeld dat het hoogste aantal SNP's 32,283 (53.96%) zich in het intronische gebied zou bevinden, gevolgd door het intergene gebied 20,395 (34.09%). Slechts 777 (1.3%) werden gedetecteerd in het exonische gebied. Net als bij GIC werd het grootste aantal SNP's gedistribueerd in de intronische regio, gevolgd door de intergene en exonische regio in alle andere rundveerassen. In SAC werd bijvoorbeeld 53.87% van de SNP's (8429) voorspeld in het intronische gebied, gevolgd door het intergenische gebied 33% (5163 SNP's) en slechts 1.75% (273 SNP's) in het exonische gebied. Een vergelijkbare trend werd waargenomen voor RAC-, RSC-, KAC- en THC-rundveerassen met respectievelijk 6834 (55.63%), 11,147 (52.12%), 8429 (53.87%), 6374 (52.58%) SNP's in de intronische regio, 4186 (34.08). %), respectievelijk 8192 (38.30%), 5163 (33%), 4507 (37.18%) SNP's in het intergenische gebied en slechts 142 (1.16%), 266 (1.24%), 273 (1.75%), 123 (1.02 %) werden voorspeld in de exonische regio's (Fig. 3). Het aantal synonieme varianten geïdentificeerd in GIC, KAC, RAC, RSC, SAC en THC was respectievelijk 570, 190, 101, 172, 213 en 87. Het aantal niet-synonieme varianten dat werd gedetecteerd voor de 6 rundveerassen was daarentegen respectievelijk 165, 64, 31, 82, 53 en 30. de TS/TV verhouding waargenomen in GIC, KAC, RAC RSC SAC en THC was respectievelijk 2.55, 2.64, 2.33, 2.43, 2.51 en 2.19 (aanvullende tabel S6).
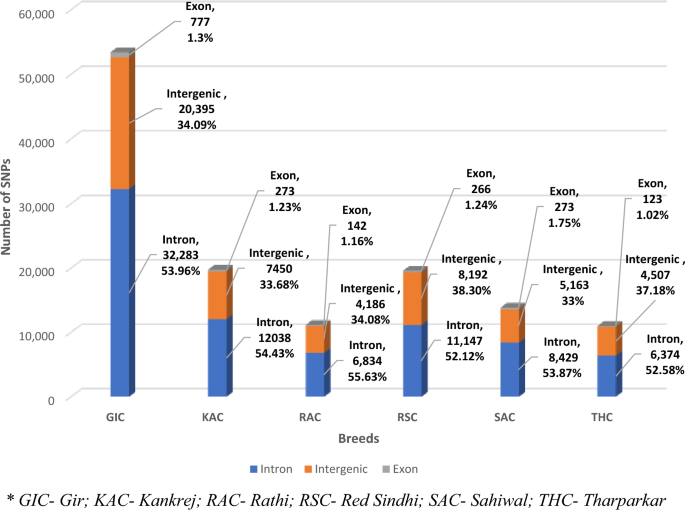
Genomische verdeling van SNP's over het genoom van zes Indiase melkveerassen.
Het aantal intergene SNP's was 4,639,873 (68.1%) en 1,676,710 (24.6%) waren intronisch. Er waren 230,365 (3.4%) SNP's binnen 5 kb stroomopwaarts en 197,827 (2.9%) stroomafwaarts van een transcriptiestartplaats; 12,428 SNP's bevonden zich in de 5' UTR en 2613 in de 3' UTR. In totaal waren 4356 SNP's gelokaliseerd op splitsingsplaatsen van 2966 genen: 142 bevonden zich op splitsingsdonorplaatsen, 142 waren splitsingsacceptorplaatsen en 4072 bevonden zich in het gebied van de splitsingsplaats. We identificeerden 45,776 SNP's die de coderende sequenties van 11,538 genen beïnvloeden. Van 221 SNP's werd voorspeld dat ze een voortijdig stopcodon veroorzaken en van 17 SNP's een toename in de coderingssequentie. Het aantal SNP's waarvan werd voorspeld dat ze niet-synoniem zijn, was 20,828. Het aantal intergene SNP's was 4,639,873 (68.1%) en 1,676,710 (24.6%) waren intronisch. Er waren 230,365 (3.4%) SNP's binnen 5 kb stroomopwaarts en 197,827 (2.9%) stroomafwaarts van een transcriptiestartplaats; 12,428 SNP's bevonden zich in de 5' UTR en 2613 in de 3' UTR. In totaal waren 4356 SNP's gelokaliseerd op splitsingsplaatsen van 2966 genen: 142 bevonden zich op splitsingsdonorplaatsen, 142 waren splitsingsacceptorplaatsen en 4072 bevonden zich in het gebied van de splitsingsplaats. Het aantal intergene SNP's was 4,639,873 (68.1%) en 1,676,710 (24.6%) waren intronisch. Er waren 230,365 (3.4%) SNP's binnen 5 kb stroomopwaarts en 197,827 (2.9%) stroomafwaarts van een transcriptiestartplaats; 12,428 SNP's bevonden zich in de 5' UTR en 2613 in de 3' UTR. In totaal waren 4356 SNP's gelokaliseerd op splitsingsplaatsen van 2966 genen: 142 bevonden zich op splitsingsdonorplaatsen, 142 waren splitsingsacceptorplaatsen en 4072 bevonden zich in het gebied van de splitsingsplaats. We identificeerden 45,776 SNP's die de coderende sequenties van 11,538 genen beïnvloeden. Van 221 SNP's werd voorspeld dat ze een voortijdig stopcodon veroorzaken en van 17 SNP's een toename in de coderingssequentie. Het aantal SNP's waarvan werd voorspeld dat ze niet-synoniem zijn, was 20,828. Het aantal intergene SNP's was 4,639,873 (68.1%) en 1,676,710 (24.6%) waren intronisch. Er waren 230,365 (3.4%) SNP's binnen 5 kb stroomopwaarts en 197,827 (2.9%) stroomafwaarts van een transcriptiestartplaats; 12,428 SNP's bevonden zich in de 5' UTR en 2613 in de 3' UTR. In totaal waren 4,356 SNP's gelokaliseerd op splitsingsplaatsen van 2966 genen: 142 bevonden zich op splitsingsdonorplaatsen, 142 waren splitsingsacceptorplaatsen en 4072 bevonden zich in het gebied van de splitsingsplaats. We identificeerden 45,776 SNP's die de coderende sequenties van 11,538 genen beïnvloeden. Van 221 SNP's werd voorspeld dat ze een voortijdig stopcodon veroorzaken en van 17 SNP's een toename in de coderingssequentie. Het aantal SNP's waarvan werd voorspeld dat ze niet-synoniem zijn, was 20,828.
Binnen rasdiversiteit
De nucleotide-diversiteit (π) was het hoogst in THC (π = 0.458), gevolgd door RSC (π = 0.364), SAC (π = 0.363), GIC (π = 0.356), KAC (π = 0.348) en RAC (π = 0.347 ). De gemiddelde nucleotide-diversiteitswaarde was 0.373 (Tabel 3). De D-waarden van de Tajima waren negatief voor 4 rundveerassen, namelijk RSC, RAC, SAC en THC behalve voor GIC en SAC waar positieve D-waarden werden waargenomen. De hoogste negatieve D-waarde van Tajima werd waargenomen in THC (-1.194), gevolgd door RSC (− 1.088), RAC (− 0.295) en KAC (− 0.279).
De waargenomen heterozygositeit (HO) waarden varieerden van 0.464 tot 0.551 terwijl de verwachte heterozygositeit (HE) varieerde van 0.448 tot 0.535. De hoogst waargenomen heterozygositeitswaarden werden waargenomen in THC (HO = 0.551) gevolgd door RAC (HO = 0.523), RSC (HO = 0.5184), SAC (HO = 0.5180), GIC (HO = 0.499) en KAC (HO = 0.464) (tabel 4). De gemiddelde FIS (inteeltcoëfficiënt) varieert van -0.253 in THC tot 0.0513 in KAC. de FIS schatting onder de zes runderrassen was het hoogst in THC (FIS = − 0.253) gevolgd door RAC (FIS = − 0.105), terwijl de laagste FIS schatting werd waargenomen in KAC (FIS = 0.0513) gevolgd door GIC (FIS = − 0.00063). De algehele FIS analyse onthulde een overmaat aan heterozygositeit voor alle runderrassen behalve voor KAC (Tabel 4). De heterozygositeit en FIS schattingen wezen op de aanwezigheid van voldoende diversiteit binnen de zes runderrassen.
Tussen rasdiversiteit
De genetische differentiatie op basis van fixatie-index (FST) varieerde van 0.2840 tot 0.3905, wat aangeeft dat er voldoende diversiteit is tussen rassen. De grootste divergentie werd waargenomen tussen het RAC-SAC-paar (FST = 0.3905), gevolgd door RSC-RAC kweekkoppel (FST = 0.3790), RSC-SAC kweekkoppel (FST = 0.3751). De minste divergentie werd waargenomen voor het KAC-THC-raspaar (FST = 0.2840) (tabel 5). Op Neighbour Joining (NJ) gebaseerde boomconstructie werden de individuele dieren van 6 rundveerassen gegroepeerd volgens hun rasrelaties, waarbij GIC en RSC het meest diverse ras waren van de 6 bestudeerde runderrassen. De fylogenetische relatie op individueel niveau wordt getoond in Fig. 4. De raswijze NJ-boom afgebeeld in Fig. 5, min of meer bevestigd met de individuele niveauboom. Bovendien werd een op UPGMA gebaseerde fylogenetische boom geconstrueerd op rasniveau met behulp van het "phangorn" -pakket in het R-platform met 100 bootstrap-waarden. De bootstrap-waarden van elk knooppunt waren bijna 100%, wat wijst op een hoge robuustheid van de geconstrueerde boom. De op UPGMA gebaseerde fylogenetische boom weerspiegelde een vergelijkbare genetische verwantschap, zoals onthuld door op NJ gebaseerde genetische differentiatie (individueel en op rasniveau), waarbij GIC en RSC de meest verschillende rassen bleken te zijn. GIC verscheen op het hoofdknooppunt en clusterde als één groep, terwijl de andere populaties twee groepen vormden met RSC geclusterd op één knooppunt en RAC, THC, SAC en KAC vormden andere subclusters (Fig. 6).
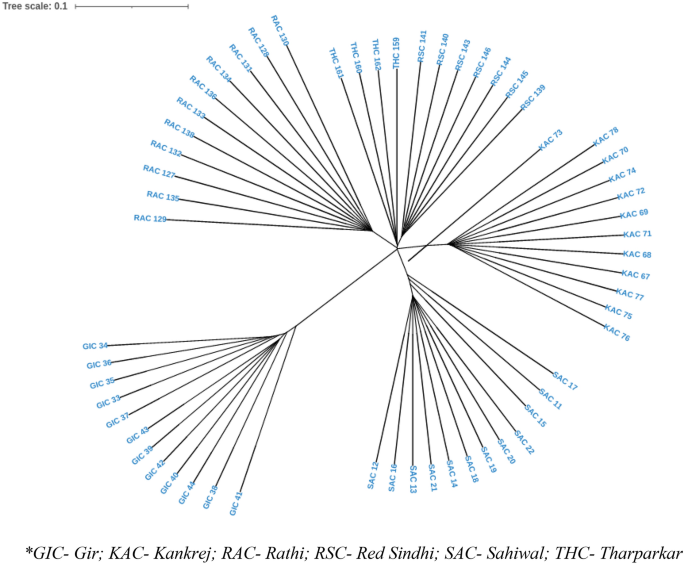
Neighbour-Joining-gebaseerde fylogenetische groepering van 58 dieren van zes Indiase melkveerassen met behulp van Tassel-software.
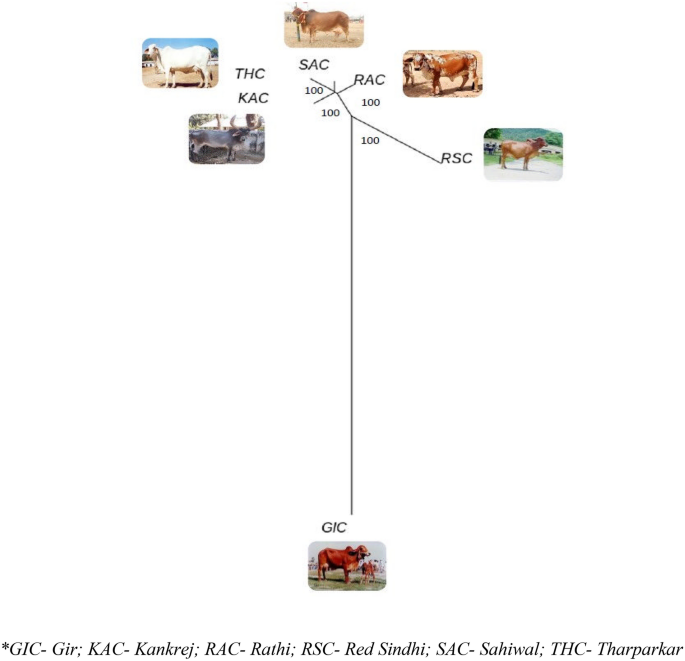
Op buurlanden gebaseerde groepering van 6 Indiase melkveerassen met behulp van het "phangorn" -pakket van het R-platform.
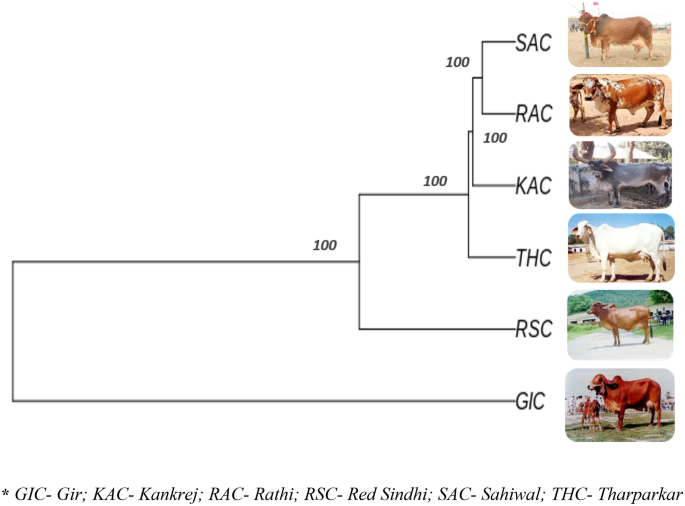
Op UPGMA gebaseerde fylogenetische groepering van zes Indiase melkrassen met behulp van het "phangorn" -pakket van het R-platform.
Analyse van de bevolkingsstructuur
De vermengingsanalyse werd uitgevoerd door het genoom van elk individu in een vooraf gedefinieerd cluster te verdelen. De analyse werd uitgevoerd bij K = 3, 4, 5 en 6 (Fig. 7). De individuen konden niet worden gegroepeerd op K = 3 volgens hun respectievelijke ras. Alleen GIC kon duidelijk worden onderscheiden, terwijl de individuen van KAC en SAC als één groep verschijnen en RAC, THC en RSC bij elkaar zijn geclusterd, wat hun gedeelde afkomst aangeeft. Bij K = 4, en zelfs bij K = 5, clusterden THC, RAC en RSC samen, wat hun sterke gedeelde voorouders aangeeft, terwijl alle andere individuen clusterden in hun eigen respectieve ras. De beste K in de analyse van de populatiestructuur is K = 6, waarbij bijna alle dieren werden gegroepeerd naar hun respectieve ras, wat duidelijk hun aparte voorouders aangeeft, met uitzondering van RSC en THC die nog steeds samen clusterden. De genetische verwantschap tussen RSC en THC zou kunnen worden onthuld door verdere diepgaande studies en door het aantal monsters te vergroten.
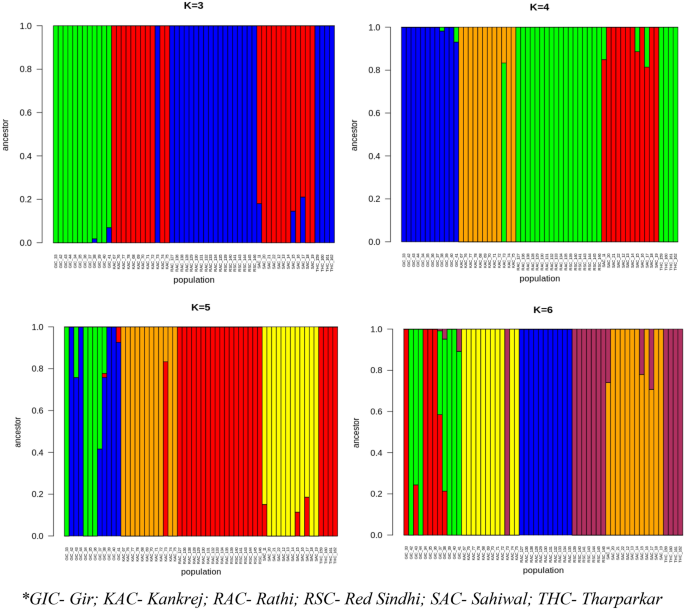
Bijmenganalyse uitgaande van 3 ≤ K ≤ 6.
De op PCA gebaseerde analyse clusterde ook 6 runderrassen afzonderlijk en versterkt het feit dat dit verschillende runderrassen zijn (aanvullend Fig. S1). Individuen van KAC werden gegroepeerd in één kwadrant, terwijl individuen van SAC RAC-, THC- en RSC-rundveerassen in een ander kwadrant vallen. Individuen van het GIC-runderras verschenen als een afzonderlijke populatie.
- Door SEO aangedreven content en PR-distributie. Word vandaag nog versterkt.
- EVM Financiën. Uniforme interface voor gedecentraliseerde financiën. Toegang hier.
- Quantum Media Groep. IR/PR versterkt. Toegang hier.
- PlatoAiStream. Web3 gegevensintelligentie. Kennis versterkt. Toegang hier.
- Bron: https://www.nature.com/articles/s41598-023-32418-6

