품질 관리, 정렬 및 SNP 호출
58개의 토착 소 품종에 속하는 XNUMX명의 개체에 대한 ddRAD 시퀀싱 기반 유전자형 분석; Gir, Sahiwal, Tharparkar, Rathi, Red Sindhi 및 Kankrej 소의 지리적 및 생태학적 분포(그림. 1) 보충 표에 제시된 각 개인의 동물 ID 및 성별과 함께 생산 목적, 코트 색상, 대표 농기후 지역, 번식지, 각 번식지의 지리적 좌표를 포함합니다. S1; 품종당 138.59만 판독 및 동물당 23만 판독에 해당하는 2.2억 138.58만 원시 판독이 발생했습니다. 읽기 품질 및 어댑터 제거를 기반으로 한 초기 필터링 후 대부분의 읽기(99.9억 XNUMX만 읽기, XNUMX%)가 유지되었습니다(보충 표 S2). 높은 비율의 읽기(94.53%)가 보스 토러스 (ARS-UCD1.2) 참조 어셈블리(보충 표 S2). 본 연구에서는 다양한 소 품종에 걸친 SNP만을 분석하기 위한 노력을 기울였기 때문에 후속 분석에서 다른 모든 변이체는 고려하지 않았다. 6개 소 품종의 SNP 수는 개별 변형 호출 후 8,42,768개에서 3,81,966개 사이였습니다. 최대 SNP 수는 SAC(8,42,768)에서 관찰되었으며, GIC(8,34,780), KAC(8,10,279), RAC(8,05,020), RSC(6,72,632) 및 THC(3,81,966) 순이었습니다. (테이블 1). 6개의 소 품종에 걸쳐 결합된 데이터 세트는 총 43,47,445개의 SNP를 생성했습니다. 그런 다음 VCF 파일을 단계별로 처리하여 품질이 낮은 SNP를 필터링했습니다. 먼저, SNP는 판독 깊이 2(RD 2), 판독 깊이 5(RD 5) 및 판독 깊이 10(RD 10)에서 필터링되었습니다. 추가 분석을 위해 RD 9,82,174에서 확인된 5개의 SNP 데이터 세트를 후속 분석에 사용했습니다(표 1). 낮은 적용 범위(RD < 5)에 존재하는 모든 SNP는 데이터 세트에서 제거되었습니다. 5의 RD에서 식별된 SNP는 누락된 유전자형의 비율, 소수 대립유전자 빈도 및 HWE(Hardy Weinberg Equilibrium)와 같은 다양한 기준을 사용하여 추가로 필터링되었습니다. 일련의 필터링 결과 총 84,027개의 고품질 SNP가 생성되었습니다. 필터링 후 품종 간 SNP의 수는 상당히 다양했습니다. SNP의 개수는 GIC(34,743)에서 가장 많았고 RSC(13,092), KAC(12,812), SAC(8956), THC(7356), RAC(7068) 순이었다(표 2).
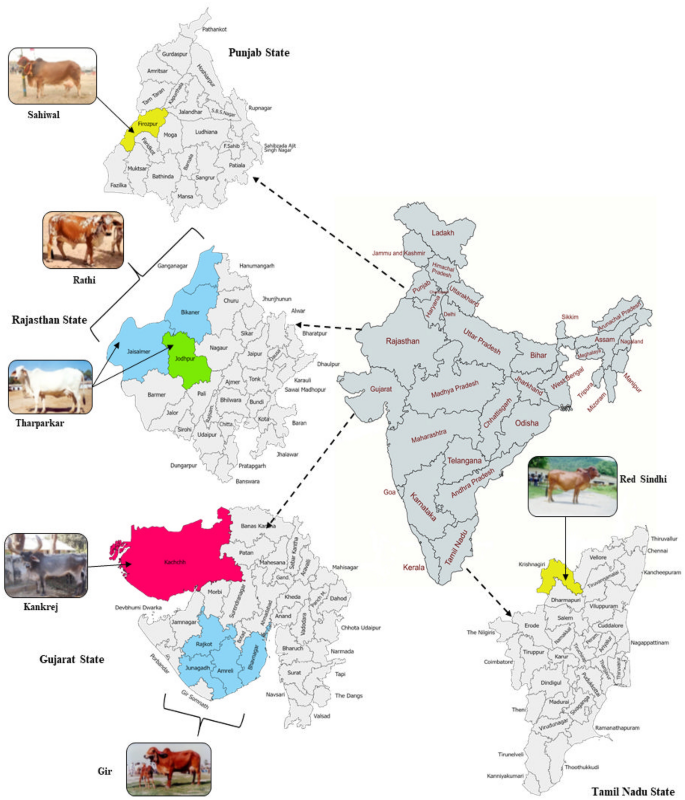
이 연구에 포함된 XNUMX가지 소 품종의 지리적 분포(지도는 웹 사이트를 사용하여 생성됨 지도 차트 https://www.mapchart.net/ 및 페인트 맵 https://paintmaps.com/).
변형의 기능 주석
모든 6가지 우유 품종의 병합된 고품질 SNP 데이터 세트는 다음과 같이 주석 처리되었습니다. 보스 토러스 (ARS-UCD1.2) 참조 게놈. 게놈에서의 그들의 분포와 관련하여, 많은 수의 주석이 달린 SNP가 인트론 영역(41,372 SNP, 53.87%)에 있을 것으로 예측되었고, 그 다음 유전자간 영역(26,834 SNP, 34.94%)에 있을 것으로 예측되었습니다. 엑소닉 영역에 분포된 SNP는 948개(1.23%)뿐이었습니다. 또한, 3497Kb 영역 상류에 4.55개의 SNP(5%)가 있고, 전사 개시 부위의 하류에 3661개의 SNP(4.77%)가 있었다. 또한 분석 결과 93'UTR에 0.121개의 SNP(5%), 293'UTR 영역에 0.38개의 SNP(3%)가 있었습니다. 총 8개의 SNP(0.01%)가 조기 정지 코돈을 유발할 것으로 예측되었으며 또한 확인되었습니다(그림 XNUMX). 2).
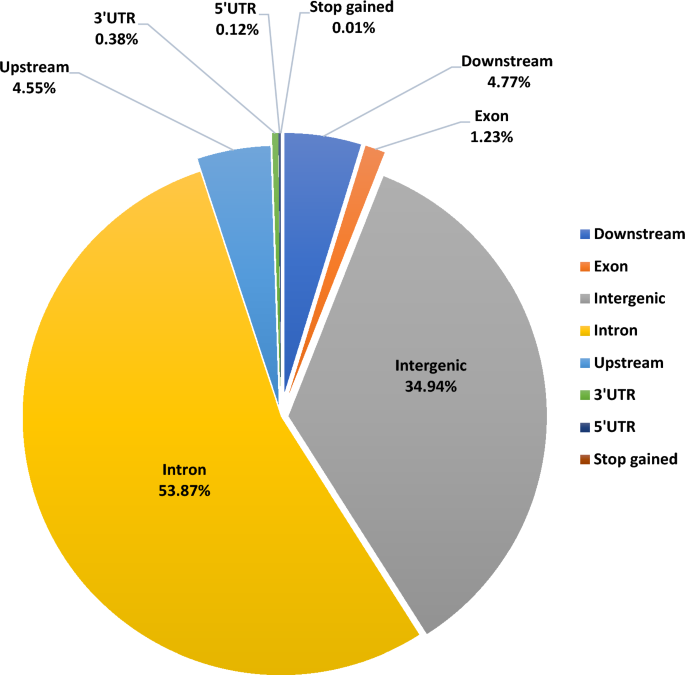
모든 품종에 대한 게놈 분포와 관련하여 SNP의 전체 분할.
단백질 코딩 유전자에 대한 SNP의 영향을 기반으로 SNP는 높은 영향(10 SNP; 0.01%), 중간 영향(298 SNP; 0.39%) 및 낮은 영향(697 SNP; 0.91%)으로 분류되었습니다. . 대부분의 SNP(75,801; 98.69%)가 수정자로 확인되었습니다(보충 표 S3). 또한, 높은 비율의 SNP(65.74%)는 본질적으로 침묵했고, 그 다음으로 미스센스(33.37%) 및 넌센스(0.89%)가 있었으며, 평균 미스센스/침묵 비율은 0.507이었습니다(보충 표 S4). 또한, 본 연구에서 확인된 치환된 모든 유전자형 중에서 C/T와 G/A 유전자형이 우세한 반면, A/T 유전자형은 가장 낮은 비율을 보였다(보충표 S5). 개별 품종에 대한 주석 결과는 그림 XNUMX에 요약되어 있습니다. 3 보조 표 S6. GIC에서 가장 많은 수의 SNP 32,283개(53.96%)가 인트론 영역에 있을 것으로 예측되었고, 그 다음이 유전자간 영역 20,395개(34.09%)에 있을 것으로 예측되었습니다. 777개(1.3%)만이 엑소닉 영역에서 검출되었습니다. GIC와 유사하게, 가장 많은 수의 SNP가 다른 모든 소 품종에서 인트론 영역에 분포하고 그 다음이 유전자간 및 엑손 영역에 분포했습니다. 예를 들어, SAC에서 SNP의 53.87%(8429)가 인트론 영역에서 예측되었고, 그 다음으로 유전자간 영역이 33%(5163 SNP), 엑손 영역에서 1.75%(273 SNP)만이 예측되었습니다. 유사한 경향이 RAC, RSC, KAC 및 THC 소 품종에 대해 관찰되었으며, 인트론 영역에서 각각 6834(55.63%), 11,147(52.12%), 8429(53.87%), 6374(52.58%) SNP, 4186(34.08) %), 8192개(38.30%), 5163개(33%), 4507개(37.18%) SNP는 유전자간 영역에서 각각 142개(1.16%), 266개(1.24%), 273개(1.75%), 123개(1.02 %)는 엑소닉 영역에서 예측되었다(도 XNUMX). 3). GIC, KAC, RAC, RSC, SAC 및 THC에서 확인된 동의 변이체의 수는 각각 570, 190, 101, 172, 213 및 87개였습니다. 한편, 6종 소 품종에 대해 검출된 비동의 변이의 수는 각각 165개, 64개, 31개, 82개, 53개, 30개였다. 티S/TV GIC, KAC, RAC RSC SAC 및 THC에서 관찰된 비율은 각각 2.55, 2.64, 2.33, 2.43, 2.51 및 2.19였습니다(보충 표 S6).
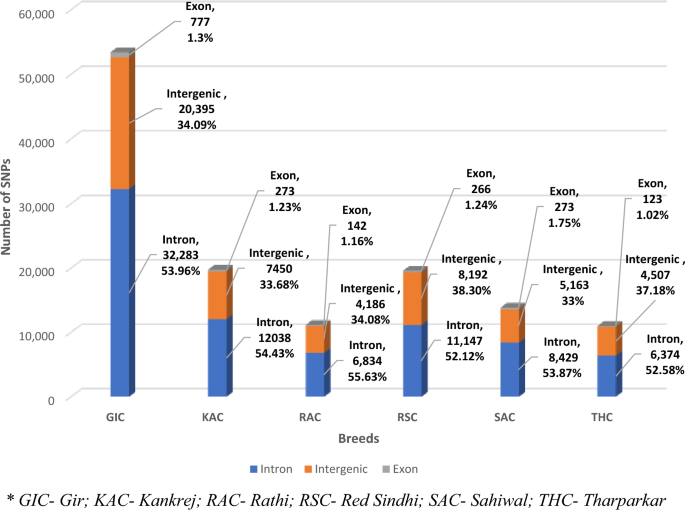
XNUMX개의 인도 젖소 품종의 게놈에 걸친 SNP의 게놈 분포.
intergenic SNP의 수는 4,639,873개(68.1%)이고 1,676,710개(24.6%)는 intronic이었다. 230,365개(3.4%)의 SNP가 전사 시작 부위의 5kb 업스트림 내에 위치하고 다운스트림에 197,827개(2.9%)가 있습니다. 12,428개의 SNP는 5' UTR에, 2613개는 3' UTR에 위치했습니다. 총 4356개의 SNP가 2966개 유전자의 스플라이스 부위에 위치하였다: 142개는 스플라이스-공여체 부위에 있었고, 142개는 스플라이스-수용체 부위였으며 4072개는 스플라이스 부위 영역 내에 있었다. 우리는 45,776개 유전자의 코딩 서열에 영향을 미치는 11,538개의 SNP를 확인했습니다. 221개의 SNP가 조기 정지 코돈을 야기할 것으로 예측되었고 17개가 코딩 서열에서 이득을 야기했습니다. 동의어가 아닌 것으로 예측된 SNP의 수는 20,828개였습니다. intergenic SNP의 수는 4,639,873개(68.1%)이고 1,676,710개(24.6%)는 intronic이었다. 230,365개(3.4%)의 SNP가 전사 시작 부위의 5kb 업스트림 내에 위치하고 다운스트림에 197,827개(2.9%)가 있습니다. 12,428개의 SNP는 5' UTR에, 2613개는 3' UTR에 위치했습니다. 총 4356개의 SNP가 2966개 유전자의 스플라이스 부위에 위치하였다: 142개는 스플라이스-공여체 부위에 있었고, 142개는 스플라이스-수용체 부위였으며 4072개는 스플라이스 부위 영역 내에 있었다. intergenic SNP의 수는 4,639,873개(68.1%)이고 1,676,710개(24.6%)는 intronic이었다. 230,365개(3.4%)의 SNP가 전사 시작 부위의 5kb 업스트림 내에 위치하고 다운스트림에 197,827개(2.9%)가 있습니다. 12,428개의 SNP는 5' UTR에, 2613개는 3' UTR에 위치했습니다. 총 4356개의 SNP가 2966개 유전자의 스플라이스 부위에 위치하였다: 142개는 스플라이스-공여체 부위에 있었고, 142개는 스플라이스-수용체 부위였으며 4072개는 스플라이스 부위 영역 내에 있었다. 우리는 45,776개 유전자의 코딩 서열에 영향을 미치는 11,538개의 SNP를 확인했습니다. 221개의 SNP가 조기 정지 코돈을 야기할 것으로 예측되었고 17개가 코딩 서열에서 이득을 야기했습니다. 동의어가 아닌 것으로 예측된 SNP의 수는 20,828개였습니다. intergenic SNP의 수는 4,639,873개(68.1%)이고 1,676,710개(24.6%)는 intronic이었다. 230,365개(3.4%)의 SNP가 전사 시작 부위의 5kb 업스트림 내에 위치하고 다운스트림에 197,827개(2.9%)가 있습니다. 12,428개의 SNP는 5' UTR에, 2613개는 3' UTR에 위치했습니다. 총 4,356개의 SNP가 2966개 유전자의 스플라이스 부위에 위치하였다: 142개는 스플라이스 공여자 부위에, 142개는 스플라이스-수용체 부위에, 4072개는 스플라이스 부위 영역 내에 있었다. 우리는 45,776개 유전자의 코딩 서열에 영향을 미치는 11,538개의 SNP를 확인했습니다. 221개의 SNP가 조기 정지 코돈을 야기할 것으로 예측되었고 17개가 코딩 서열에서 이득을 야기했습니다. 동의어가 아닌 것으로 예측된 SNP의 수는 20,828개였습니다.
품종 다양성 내에서
뉴클레오티드 다양성(π)은 THC(π = 0.458)에서 가장 높았고 RSC(π = 0.364), SAC(π = 0.363), GIC(π = 0.356), KAC(π = 0.348) 및 RAC(π = 0.347) 순이었습니다. ). 평균 뉴클레오티드 다양성 값은 0.373(표 3). Tajima의 D 값은 4가지 소 품종 즉, RSC, RAC, SAC 및 THC에 대해 음수였으며 GIC 및 SAC는 양의 D 값이 관찰되었습니다. THC(-1.194)에서 가장 높은 음의 Tajima D 값이 관찰되었고 RSC(-1.088), RAC(-0.295) 및 KAC(-0.279)가 그 뒤를 이었습니다.
관찰된 이형접합체(HO) 값의 범위는 0.464에서 0.551인 반면 예상되는 이형접합체(HE) 범위는 0.448에서 0.535입니다. 관찰된 가장 높은 이형접합성 값은 THC(HO = 0.551)에 이어 RAC(HO = 0.523), RSC(HO = 0.5184), SAC(HO = 0.5180), GIC(HO = 0.499) 및 KAC(HO = 0.464) (표 4). 평균 FIS (근친 교배 계수)의 범위는 THC에서 -0.253에서 KAC에서 0.0513입니다. 더에프IS XNUMX종의 소 품종 중 THC(FIS = - 0.253) RAC(FIS = - 0.105), 가장 낮은 FIS 추정치는 KAC(FIS = 0.0513) 다음에 GIC(FIS = - 0.00063). 전체 FIS 분석 결과 KAC를 제외한 모든 소 품종에 대해 과도한 이형 접합체가 나타났습니다 (표 4). 이형접합체와 FIS 추정치는 XNUMX종의 소 품종 내에서 충분한 다양성이 존재함을 나타냅니다.
품종 다양성 사이
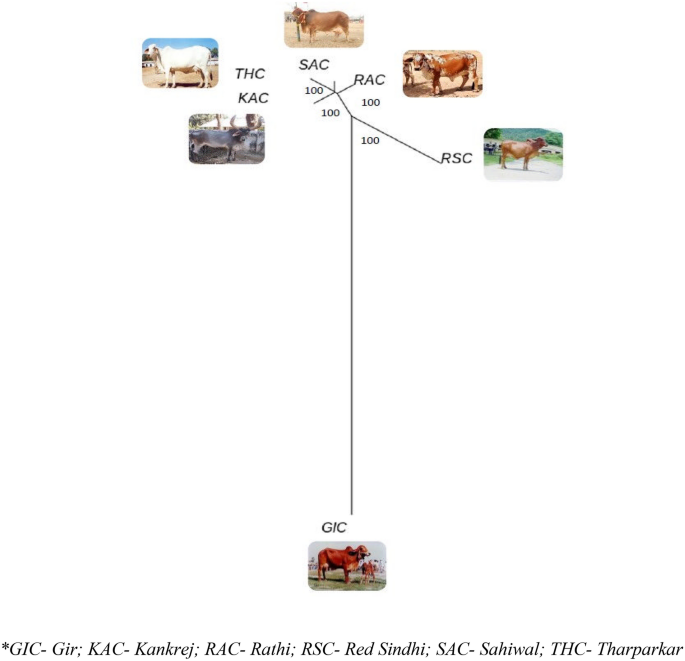
고정 지수(FST) 범위는 0.2840에서 0.3905로 품종 다양성이 충분함을 나타냅니다. RAC-SAC 쌍(FST = 0.3905), RSC-RAC 품종 쌍(FST = 0.3790), RSC-SAC 품종 쌍(FST = 0.3751). KAC-THC 품종 쌍(FST = 0.2840) (표 5). NJ(Neighbor Joining) 기반 트리를 구축하고 6개의 연구된 소 품종 중에서 가장 다양한 품종인 GIC 및 RSC와 품종 제휴에 따라 6가지 소 품종의 개별 동물을 그룹화했습니다. 개별 수준의 계통 발생 관계는 그림 XNUMX에 나와 있습니다. 4. 그림에 묘사된 번식 현명한 NJ 나무. 5, 개별 레벨 트리로 어느 정도 확증됩니다. 또한 UPGMA 기반 계통수는 100개의 부트스트랩 값으로 R 플랫폼의 "phangorn" 패키지를 사용하여 품종 수준에서 구성되었습니다. 각 노드의 부트스트랩 값은 100%에 가까웠으며 구성된 트리의 견고성이 높음을 나타냅니다. UPGMA 기반 계통발생수는 GIC와 RSC가 가장 뚜렷한 품종으로 나타난 NJ 기반 유전적 분화(개체별 및 품종 수준)에 의해 밝혀진 것과 유사한 유전적 관계를 반영했습니다. GIC는 주요 노드에 나타나고 하나의 그룹으로 클러스터링되는 반면 다른 개체군은 RSC가 하나의 노드에 클러스터되고 RAC, THC, SAC 및 KAC가 다른 하위 클러스터를 형성하는 두 그룹을 형성했습니다(그림 XNUMX). 6).
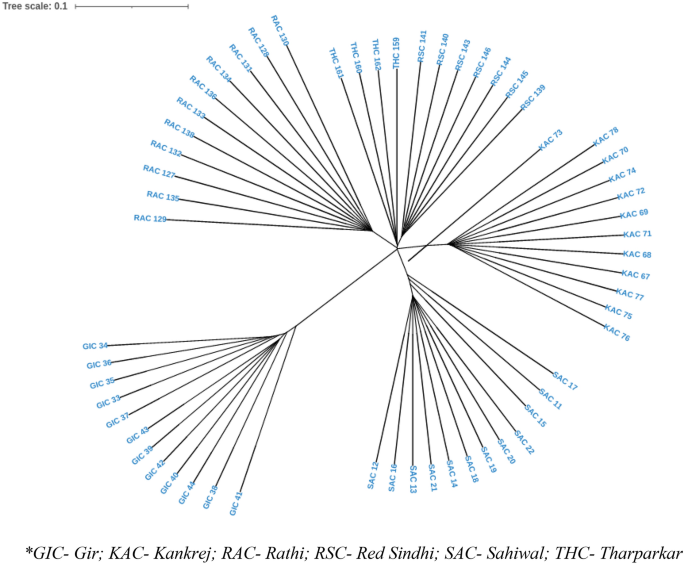
Tassel 소프트웨어를 사용하여 58개의 인도 젖소 품종의 XNUMX마리 동물의 이웃 결합 기반 계통 발생 그룹화.
R 플랫폼의 "phangorn" 패키지를 사용하여 6개의 인도 젖소 품종의 이웃 결합 기반 그룹화.
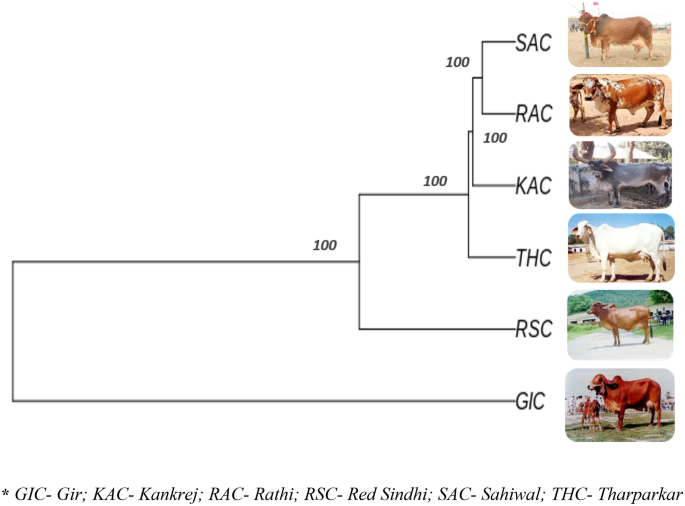
R 플랫폼의 "phangorn" 패키지를 사용하여 XNUMX개의 인도 우유 품종의 UPGMA 기반 계통 발생 그룹화.
인구 구조 분석
혼합물 분석은 각 개인의 게놈을 미리 정의된 클러스터로 분할하여 수행되었습니다. 분석은 K = 3, 4, 5 및 6에서 수행되었습니다(그림 XNUMX). 7). 개체는 각각의 품종에 따라 K = 3에서 그룹화할 수 없습니다. KAC와 SAC의 개체가 하나의 그룹으로 나타나고 RAC, THC 및 RSC가 함께 클러스터되어 공유 조상을 나타내는 반면 GIC만 명확하게 구분할 수 있습니다. K = 4, 심지어 K = 5에서도 THC, RAC 및 RSC는 강력한 공유 조상을 나타내는 함께 모여 있는 반면, 다른 모든 개인은 각자의 품종으로 모여 있습니다. 개체군 구조 분석에서 가장 좋은 K는 K = 6이며, 여기서 거의 모든 동물이 각각의 품종으로 그룹화되어 여전히 함께 모여 있는 RSC와 THC를 제외하고 서로 다른 조상을 명확하게 나타냅니다. RSC와 THC 사이의 유전적 근접성은 추가 심층 연구와 샘플 수 증가를 통해 밝혀질 수 있습니다.
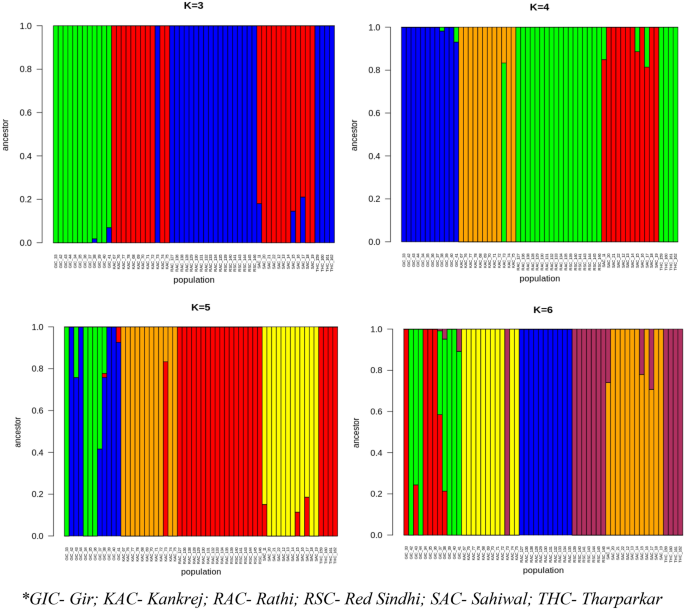
3 ≤ K ≤ 6으로 가정한 혼합물 분석.
PCA 기반 분석은 또한 6개의 소 품종을 개별적으로 클러스터링했으며 이들이 별개의 소 품종이라는 사실을 강화합니다(보충 그림 XNUMX). S1). KAC의 개체는 하나의 사분면에 함께 그룹화되었으며 SAC RAC, THC 및 RSC 소 품종의 개체는 다른 사분면에 속합니다. GIC 소 품종의 개체는 별개의 개체군으로 나타났습니다.
- SEO 기반 콘텐츠 및 PR 배포. 오늘 증폭하십시오.
- EVM 금융. 탈중앙화 금융을 위한 통합 인터페이스. 여기에서 액세스하십시오.
- 퀀텀미디어그룹. IR/PR 증폭. 여기에서 액세스하십시오.
- PlatoAiStream. Web3 데이터 인텔리전스. 지식 증폭. 여기에서 액세스하십시오.
- 출처: https://www.nature.com/articles/s41598-023-32418-6

